進行性核上性眼肌麻痹(附1例報道)
2017-08-07 05:24:08郭樹晨李承晏
卒中與神經疾病 2017年3期
郭樹晨 李承晏
?
進行性核上性眼肌麻痹(附1例報道)
郭樹晨 李承晏
目的 報道1例少見的進行性核上性眼肌麻痹(progressive supranuclear palsy, PSP)治療可能有效的患者。方法 回顧性分析1例臨床擬診為PSP患者的診斷及治療,并復習文獻。結果 患者臨床表現為認知功能障礙、運動障礙、垂直性眼肌麻痹、飲水嗆咳;顱腦MRI示額顳葉皮質明顯萎縮、軸位顯示中腦“牽牛花征”、矢狀位顯示中腦被蓋部“蜂鳥征”。給予大劑量美多芭、珂丹、輔酶Q10等治療后患者癥狀明顯好轉。結論 大劑量的美多芭(0.75 g/d)、珂丹(0.3 g/d)可能改善PSP患者的臨床癥狀,但應結合患者的實際情況,注意其相關的不良反應,本例患者對大劑量的美多芭、珂丹耐受性較好,未出現不良反應。
核上性眼肌麻痹 進行性 臨床表現 診斷 治療
進行性核上性眼肌麻痹(progressive supranuclear palsy PSP)是一種由Tau蛋白病變引起的散發性神經退行性疾病,其患病率約5~6.5/10萬[1],從出現癥狀到死亡的平均時間為7年[1]。典型的臨床表現為垂直性凝視麻痹、姿勢不穩定、容易向后摔倒、皮層下癡呆、構音障礙及吞咽困難、帕金森樣癥狀。本研究結合文獻對1例PSP患者的臨床資料進行分析總結,現報道如下。
1 臨床資料
患者,男,65歲, 2012年12月無明顯誘因出現記憶力下降,主要表現為記不住剛放的東西在什么地方、言語減少、經常一個人發呆,遂至醫院就診,診斷為“老年癡呆”給予安理申(具體用量不詳)口服治療,癥狀未見明顯緩解。2013年下半年開始出現抬頭困難、走路前傾、坐下后不能站立、經常向后摔倒,但生活尚能自理。2014年5月患者認知障礙加重,完全不能言語,走路向后摔倒的次數增多,同時左下肢開始抖動,2014年8月再到某醫院就診,診斷為“老年癡呆”給予森福羅、尼麥角林、歐來寧膠囊治療,癥狀未見好轉。11月再次至某醫院就診,診斷為“老年癡呆、帕金森氏綜合征”,停用森福羅,給予美多芭0.125 g Tid治療,患者癥狀無明顯改善,但也未加重。2015年2月出現飲水嗆咳, 3月完全不能行走,一直在醫院行康復治療,為求進一步治療,門診以“認知障礙原因待查”收入本科。既往有胃潰瘍、高脂血癥及腎結石病史,2015年2月因吸入性肺炎住院治療,2015年因摔倒形成硬膜下血腫,未給予特殊治療。家族中無類似疾病。查體:神志清楚、不語,查體欠合作,認知力、計算力、記憶力明顯下降,無法用MMSE或MOCA量表測量。雙側瞳孔等大等圓,直徑約3 mm,光反射靈敏,眼球上視不能,輻輳反射消失,未見明顯眼震,雙側鼻唇溝等稱,伸舌不配合,咽反射消失,雙上肢肌力為4級,雙下肢肌力為4級,軀干及左下肢肌張力增高,左下肢不自主抖動,無肌肉萎縮,左側腱反射活躍,雙側Babinskin征陽性,腦膜刺激征陰性。深淺復合感覺不配合,心肺腹未見明顯異常。輔助檢查:血常規、貧血五項、甲狀腺功能、凝血功能未見明顯異常,生化及電解質示尿酸663 umol/L(149~407),甘油三酯3.55 mmol/L(0.56~1.7),葡萄糖6.56 mmol/L(3.5~6.1),糖化血紅蛋白7.1%(3.5~6.5),餐后2 h血糖13.96 mmol/L。四肢肌電圖示雙下肢周圍神經損害(感覺受累為主)。顱腦MRI示雙側額顳葉皮質明顯萎縮,軸位中腦萎縮呈“牽牛花征”,第三、四腦室和中腦環池擴大,殼核和基底節T2加權像高信號;正中矢狀位中腦被蓋部萎縮呈“蜂鳥征”(圖1~4);顱腦SWAN示雙側黑質信號強度、大小等稱。入院后給予大劑量的美多芭(0.75 g/d)、珂丹(0.3 g/d)、輔酶Q10等治療后患者行走狀態較前明顯好轉,精神狀態也明顯好轉,說話較前明顯增多。
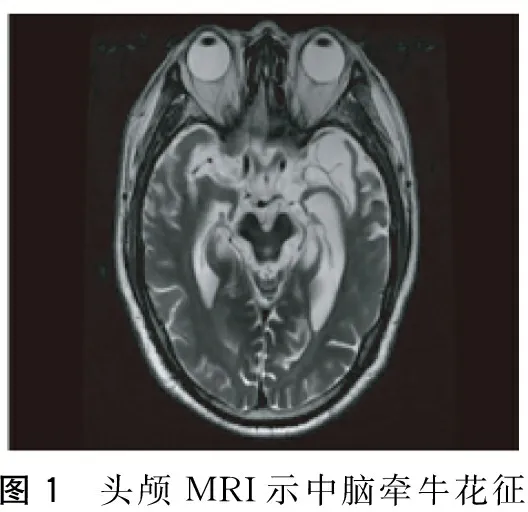
圖1 頭顱MRI示中腦牽牛花征
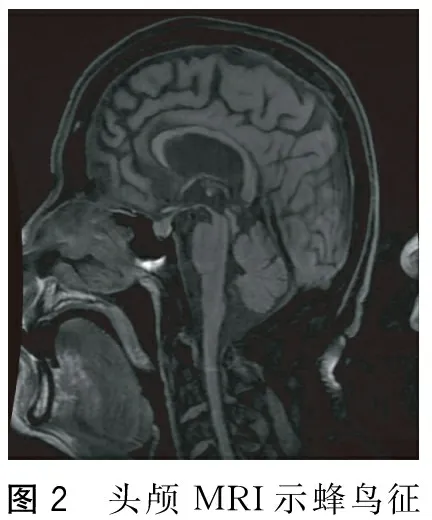
圖2 頭顱MRI示蜂鳥征

圖3 頭顱MRI示第三腦室擴大、顳葉萎縮、外側裂增寬
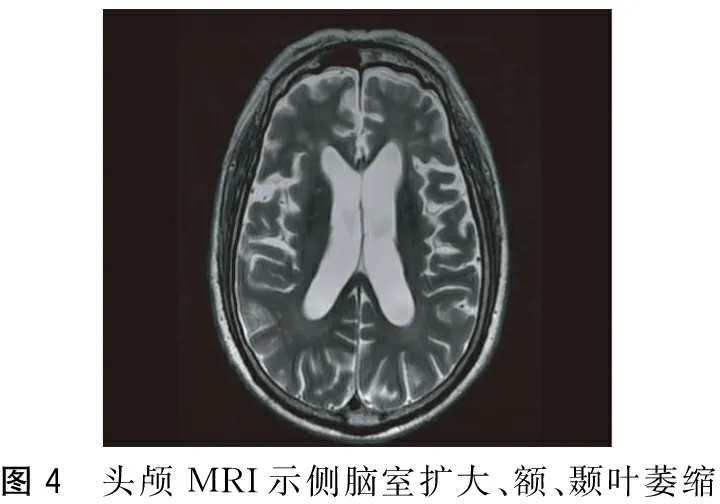
圖4 頭顱MRI示側腦室擴大、額、顳葉萎縮
2 討 論
PSP是一種散發的、成年起病的以Tau蛋白病變為病理特征的運動障礙性疾病,起病隱襲,呈進行性加重。此患者發病年齡為65歲、病情逐漸進展、有垂直性眼肌麻痹同時在疾病的第1年出現姿勢不穩定經常摔倒,符合1996年美國國立神經系統疾病與腦卒中研究所(NINDS)與進行性核上性麻痹學會(SPSP)聯合推薦并于2003年予以修訂的PSP診斷標準[2-3]中擬診PSP的入選標準,即40或40歲以后發病、病情逐漸進展、垂直性向上或向下核上性凝視麻痹。本例患者疾病的首發癥狀為認知障礙,表現為淡漠、言語減少,在疾病的第2年出現構音障礙,在病程的第3年出現飲水嗆咳,符合NINDS-PSP診斷標準中擬診PSP的支持標準中的早期出現吞咽困難和構音障礙、早期出現認知障礙如淡漠、抽象思維能力減弱、言語不流暢。本例患者從未有腦炎病史、無異己肢體綜合征的表現、在疾病的過程中未出現過明顯的小腦癥狀、無自主神經功能失調(明顯低血壓和排尿障礙)、無基底核和腦干梗死等腦部結構損害的神經放射學依據,符合NINDS-PSP診斷標準中擬診PSP的排除標準。顱腦MRI中軸位T2加權像上中腦萎縮呈“牽牛花征”、矢狀位T1加權像上呈“蜂鳥征”對預測PSP患者的病理有高度的特異性[4]。本例患者MRI有明顯的“牽牛花征”、“蜂鳥征”及三、四腦室和中腦環池擴大征。患者多次血糖監測及糖化血紅蛋白檢查提示糖尿病,可解釋其肌電圖表現,PSP可伴有錐體束征,可解釋患者雙側Babinskin征陽性體征。因此,結合其診斷的必備條件、支持條件及排除標準、特征性的影像學表現,本例患者擬診為PSP。
PSP的臨床癥狀多變,基于Tau蛋白病理學上分布的不同主要有以下幾種表型[5],即(1)PSP的Richard綜合征(PSP-RS) 臨床特征:垂直性核上性眼肌麻痹、嚴重的姿勢不穩定早期易摔倒、假性球麻痹、軸性肌強直、對稱性的、左旋多巴抵抗性運動不能及認知障礙,也可出現復視、畏光及眼瞼運動不能等其他的非典型的眼睛體征。PSP-RS早期最常出現的是姿勢不穩定伴摔倒,大部分患者在疾病的第1年出現。假性球麻痹特征包括吞咽困難和構音障礙,是PSP的支持性診斷。PSP-RS有一系列典型的認知障礙,額葉功能障礙尤其是行為和性格的改變,在PSP-RS中始終可見,并且可能在疾病的早期出現。淡漠比去抑制更常見,執行功能障礙、語言流利性減低在PSP-RS額葉障礙的患者中也很常見;(2)以帕金森氏綜合征為主要表現的PSP(PSP-P):主要表現為運動遲緩及非對稱性錐體外系強直及疾病早期階段應用左旋多巴有效,震顫、摔倒及眼睛運動障礙經常出現在疾病的晚期甚至從不出現。疾病的早期很少出現認知障礙,一些患者早期可出現吞咽困難和構音障礙。其他還包括單純性運動不能伴步態凍結(PSP-PAGF)、皮質基底節綜合征型PSP(PSP-CBS)、主要表現為說話或/和語言功能障礙的PSP(PSP-AOS和PSP-PNFA)、以小腦性共濟失調為主要表現的PSP(PSP-C)、以原發性脊髓側索硬化為主要表現的PSP(PSP-PLS)等7種類型。根據以上分型,此患者較符合“PSP-RS”。由于PSP的臨床表現各異,同時缺乏特異性檢查手段,臨床需與額顳葉癡呆、帕金森病、多系統萎縮等相鑒別。本例患者最初誤診為“老年癡呆”后被誤診為“帕金森病”。
PSP目前無特效治療。多巴胺替代治療只能暫時輕度緩解一些癥狀,有時會獲得一些益處[6],值得進行嘗試性治療。本例患者在給予大劑量的美多芭、珂丹治療后患者癥狀明顯緩解。本例大劑量美多芭、珂丹治療有效的可能原因如下:帕金森病患者運用多巴胺藥物有效是通過增加基底神經節的神經輸出,減弱來自丘腦-蒼白球的GABA抑制,而PSP患者基底神經節黑質、蒼白球、尾狀核、殼核、丘腦底核嚴重廣泛受損,因此對于常規劑量外源性的多巴胺藥物作用有限,但大劑量的美多芭、珂丹可能能夠進一步增加基底神經節的輸出,從而改善患者的臨床癥狀。因此,PSP在目前無特效治療的情況下可以根據患者的耐受情況,聯合應用大劑量的美多芭、珂丹,但是要注意其大劑量聯合應用的不良反應,主要包括異常的不隨意運動、意識模糊、失眠及罕見的惡心、嘔吐或心律失常、體位性低血壓。
[1] Koros C,Stamelou M. Interventions in progressive supranuclear palsy[J]. Parkinsonism Relate Disord,2016,22(Suppl 1):S93-S95.
[2] Litvan I,Agid Y,Calne D,et al.Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop[J].Neurology,1996,47(1):1-9.
[3] LitwanI,Hauw JJ,BartkoJJ,et al.Validity and reliability of the neuropathologic preliminary criteria for progressive supranuclear palsy and related disorders[J].Neuropathol Exp Neurol,1996,55(1):97-105.
[4] Massey LA,Micallef C,Paviour DC,et al.Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy[J].Mov Disord,2012,27(14):1754-1762.
[5] Respondek G,H glinger GU. The phenotypic spectrum of progressive supranuclear palsy[J].Parkinsonism Relat Disord, 2016, 22(Suppl 1):S34-S36.
[6] Rehman HU.Progressive supranuclear palsy[J].Postgrad Med J,2000,76(8):333-336.
(2016-07-19收稿)
430060 武漢大學人民醫院神經內科[郭樹晨 李承晏(通訊作者)]
R742
A
1007-0478(2017)03-0246-03
10.3969/j.issn.1007-0478.2017.03.020
猜你喜歡
課堂內外·初中版(科學少年)(2023年10期)2023-12-10 00:43:06
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
全科護理(2022年10期)2022-12-26 21:19:15
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
保健醫苑(2022年1期)2022-08-30 08:39:40
鄉村科技(2021年33期)2021-03-16 02:26:54
國際放射醫學核醫學雜志(2021年10期)2021-02-28 08:41:58
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
獸醫導刊(2016年6期)2016-05-17 03:50:35
