摻雜對金屬-MoS2界面性質(zhì)調(diào)制的第一性原理研究?
2017-08-09 07:34:42陶鵬程黃燕周孝好陳效雙陸衛(wèi)
物理學(xué)報(bào) 2017年11期
陶鵬程黃燕周孝好?陳效雙陸衛(wèi)
1)(中國科學(xué)院上海技術(shù)物理研究所,紅外物理國家重點(diǎn)實(shí)驗(yàn)室,上海 200083)
2)(中國科學(xué)院大學(xué),北京 100049)
摻雜對金屬-MoS2界面性質(zhì)調(diào)制的第一性原理研究?
陶鵬程1)2)黃燕1)周孝好1)?陳效雙1)陸衛(wèi)1)
1)(中國科學(xué)院上海技術(shù)物理研究所,紅外物理國家重點(diǎn)實(shí)驗(yàn)室,上海 200083)
2)(中國科學(xué)院大學(xué),北京 100049)
(2016年12月27日收到;2017年3月1日收到修改稿)
采用基于密度泛函理論的第一性原理贗勢平面波方法,計(jì)算了鹵族元素?fù)诫s對金屬-MoS2界面性質(zhì)的影響,包括缺陷形成能、電子能帶結(jié)構(gòu)、差分電荷密度以及電荷布居分布.計(jì)算結(jié)果表明:鹵族元素原子傾向于占據(jù)單層MoS2表面的S原子位置;對于單層MoS2而言,鹵族元素的摻雜將在禁帶中引入雜質(zhì)能級以及導(dǎo)致費(fèi)米能級位置的移動.對于金屬-MoS2界面體系,結(jié)合Schottky-Mott模型,證明了鹵族元素的摻雜可以有效地調(diào)制金屬-MoS2界面間的肖特基勢壘高度.發(fā)現(xiàn)F和Cl原子的摻雜將會降低體系的肖特基勢壘高度.相比之下,Br和I原子的摻雜卻增大了體系的肖特基勢壘高度.通過差分電荷密度和布居分布的分析,闡明了肖特基勢壘高度的被調(diào)制是因?yàn)殡姾赊D(zhuǎn)移形成的界面偶極矩的作用導(dǎo)致.研究結(jié)果解釋了相關(guān)實(shí)驗(yàn)現(xiàn)象,并給二維材料的器件化應(yīng)用提供了調(diào)節(jié)手段.
肖特基勢壘,二硫化鉬,摻雜,密度泛函理論
1 引言
近年來,二維材料憑借其非比尋常的優(yōu)良特性,包括電學(xué)、光學(xué)、磁學(xué)、熱學(xué)以及力學(xué)特性等引起了人們的廣泛關(guān)注,其中石墨烯[1,2]、六角氮化硼[3]、過渡性金屬硫化物[4?7]以及黑磷等[8,9]都已在理論和實(shí)驗(yàn)上得到了證實(shí).以單層MoS2為代表的過渡性金屬硫化物已經(jīng)成為研究的熱點(diǎn),也正是因?yàn)閱螌覯oS2具有許多新奇的物理和化學(xué)性質(zhì)[10].塊體的MoS2具有層狀結(jié)構(gòu),層與層之間有著微弱的范德瓦耳斯相互作用,可以通過機(jī)械剝離法獲得單層的MoS2.單層MoS2有著超薄的厚度,大約為0.65 nm,表面沒有懸掛鍵[7].單層MoS2是一種直接帶隙半導(dǎo)體材料,帶隙大約為1.8 eV[11,12].據(jù)報(bào)道,以單層MoS2為材料制成的晶體管,它的電流開/關(guān)比可以達(dá)到1×108,室溫下載流子遷移率可以達(dá)到200 cm2·V?1·s?1[7].
盡管單層MoS2有著非常優(yōu)良的本征特性,但是在其真正走向應(yīng)用之前,仍然面臨著許多科學(xué)和技術(shù)問題[7,13],其中就包括如何形成有效的電極接觸.金屬和半導(dǎo)體的接觸對于成型的器件功耗產(chǎn)生著重要影響,如何將金屬和單層過渡性金屬硫化物的接觸電阻降低是一個亟待解決的問題.為了解決這個困難,文獻(xiàn)[14,15]利用密度泛函理論定性研究了存在范德瓦耳斯作用的Ti-MoS2和Au-MoS2頂部接觸體系,發(fā)現(xiàn)最常見的接觸金屬Au為單層MoS2注入電子的效率極低,并指出Ti可以作為合適的替代電極材料;文獻(xiàn)[16,17]研究了Sc,Ni,Au與多層MoS2接觸情形,發(fā)現(xiàn)功函數(shù)較大的金屬如Ni和Pt與多層MoS2接觸存在肖特基勢壘;文獻(xiàn)[5]報(bào)道Pd-WSe2的頂位接觸是一種p型接觸;文獻(xiàn)[6]報(bào)道In-WSe2的邊緣接觸可以實(shí)現(xiàn)較低的接觸電阻;文獻(xiàn)[18]基于密度泛函理論的二維合金與MoS2接觸的研究報(bào)道指出,n型摻雜的MoS2與Ti2CF2,Ti2C(OH)2接觸均為n型接觸,肖特基勢壘勢壘高度分別為0.85和0.26 eV.金屬(電極)與單層MoS2界面形成的肖特基勢壘的高低決定著器件的接觸電阻大小,通過簡單地選擇功函數(shù)較低的金屬作為接觸電極,以此來實(shí)現(xiàn)降低接觸電阻是十分困難的,因?yàn)橘M(fèi)米能級傾向于釘扎在電荷中性區(qū)或者S空位能級處,費(fèi)米能級置于導(dǎo)帶邊緣,不能影響肖特基勢壘的高度[19,20].在金屬半導(dǎo)體接觸中,可以利用重?fù)诫s的辦法實(shí)現(xiàn)低的接觸電阻,通過重?fù)诫s,可以減小肖特基勢壘的高度,在電壓控制下可以大大增加通過金屬-半導(dǎo)體結(jié)區(qū)的電流.最近,Yang等[21]利用Cl氣氛下處理,大大減小了薄層MoS2與金屬的接觸電阻,但是其中機(jī)理尚不明確.本文通過基于密度泛函理論的第一性原理對其進(jìn)行理論驗(yàn)證,我們建立了以Au原子為代表的金屬模型,通過對體系的原子進(jìn)行結(jié)構(gòu)優(yōu)化,獲得穩(wěn)定的結(jié)構(gòu),在此基礎(chǔ)上計(jì)算它們的能帶結(jié)構(gòu),驗(yàn)證實(shí)驗(yàn)的結(jié)果,給出相應(yīng)的理論解釋,并且預(yù)測其他鹵族元素?fù)诫s后對金屬和半導(dǎo)體接觸體系肖特基勢壘的影響.
2 計(jì)算方法與模型
本文所有計(jì)算均采用基于密度泛函理論的第一性原理商業(yè)軟件Material Studio軟件中CASTEP模塊進(jìn)行[22].利用超軟贗勢描述了離子實(shí)和價(jià)電子之間的相互作用,交換關(guān)聯(lián)能采用了Perdew-Burke-Ernzerhof(PBE)形式的廣義梯度近似(generalized gradient approximation,GGA).經(jīng)收斂測試后,選取的Monkhorst-Pack K點(diǎn)取樣密度為6×6×1,平面波截?cái)嗄転?10 eV,對體系進(jìn)行幾何優(yōu)化的收斂標(biāo)準(zhǔn)為:能量低于2.0×10?5eV/atom,原子間最大作用力低于0.05 eV/?,晶體內(nèi)最大應(yīng)力小于0.1 GPa,原子最大位移小于0.002 ?.這四個參數(shù)都達(dá)到后認(rèn)為優(yōu)化成功.
我們通過對MoS2體材料原胞的優(yōu)化得到了穩(wěn)定的結(jié)構(gòu),原胞的晶格常數(shù)為a=b=3.18 ?,c=14.04 ?,以此建立了3×3×1的MoS2超胞模型.常見的金屬電極為Al,Ti,Cr,Ni,Cu,Pd,Ag,In,Pt,Au.根據(jù)肖特基模型,接觸金屬具有低的功函數(shù)可實(shí)現(xiàn)n型肖特基勢壘,或者功函數(shù)高的金屬實(shí)現(xiàn)p型肖特基勢壘.但是根據(jù)金屬功函數(shù)并不能實(shí)現(xiàn)良好的金屬半導(dǎo)體接觸.對Au原子的建模,我們先對Au的原胞進(jìn)行晶格常數(shù)的優(yōu)化,得到晶胞的晶格常數(shù)a=b=c=4.19 ?,切取了Au的6層(111)面進(jìn)行a,b方向3×3的擴(kuò)胞.Au為面心立方晶體,它的(1 1 1)面沿[1 1 1]方向按照ABCABCABC···的規(guī)律排列,為了得到最穩(wěn)定的構(gòu)型,我們分別采用Au(1 1 1)的A,B,C面(對應(yīng)圖1(e)中的Au1,Au2,Au3面)與單層MoS2接觸,對這三種構(gòu)型優(yōu)化完后,計(jì)算了這三種構(gòu)型的總能,分別為?71804.33,?71802.42,?71802.52 eV,圖1(e)呈現(xiàn)的構(gòu)型總能最低,所以它是最穩(wěn)定的構(gòu)型,本文涉及的金屬-半導(dǎo)體摻雜計(jì)算都采用這種構(gòu)型.Au-MoS2(包括摻雜體系)的真空層厚度均選為30 ?,鹵族元素的摻雜選擇替位摻雜.在計(jì)算能帶時我們選取了具有高對稱性的三個點(diǎn)Γ,M,K.能帶的計(jì)算路徑均選為:Γ-M-K-Γ(見圖1(c)).
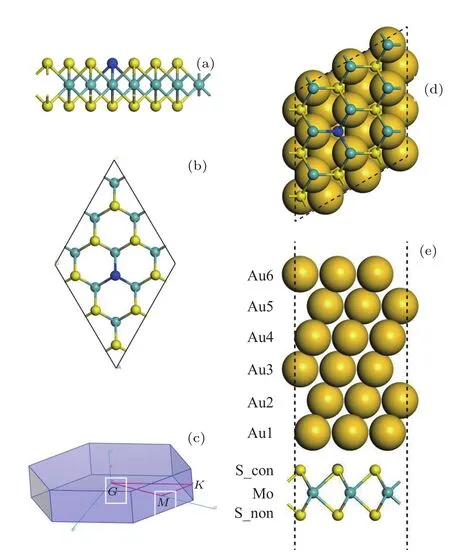
圖1 (網(wǎng)刊彩色)(a)鹵族元素?fù)诫s的MoS2側(cè)視圖;(b)鹵族元素?fù)诫s的MoS2頂視圖;(c)布里淵區(qū);(d)鹵族元素?fù)诫s的MoS2在Au(111)面的頂視圖;(e)鹵族元素?fù)诫s的MoS2在Au(111)面的側(cè)視圖Fig.1.(color online)(a)Side view of the halogendoped MoS2;(b)top view of the halogen-doped MoS2;(c)Brillouin zone;(d)top view of the halogen-doped MoS2on Au(111);(e)side view of the halogen-doped MoS2on Au(111).
3 計(jì)算結(jié)果與討論
3.1 缺陷形成能
實(shí)際情況中,材料的缺陷形成能是個十分重要的概念,通常被用來描述缺陷形成的難易程度,缺陷形成能越小說明這種缺陷越容易形成.它的定義[23]為
這里,Ef表示缺陷或摻雜的形成能;Etot(defect)為缺陷體系或者摻雜后體系的總能;E(pure)為完整材料的總能;ni表示類型為i的基體原子或者摻雜原子數(shù)目,ni>0對應(yīng)在完整體系上增加一個原子,ni<0對應(yīng)在完整體系上移走一個原子;q表示體系轉(zhuǎn)移電荷數(shù);EF表示費(fèi)米能級;EVBM(defect)表示缺陷或雜質(zhì)體系價(jià)帶頂?shù)哪芰?下面計(jì)算中取q=0進(jìn)行近似計(jì)算.
我們分別計(jì)算了超胞大小為3×3×1的單層MoS2的S和Mo空位缺陷形成能和F,Cl,Br,I替代S位摻雜的缺陷形成能.通過建立了一個1 nm3的立方晶格去計(jì)算F2(g),Cl2(g),Br2(g),I2(g),S2(g)的能量,進(jìn)而估算出F,Cl,Br,I,S原子的化學(xué)勢.此外,由于化學(xué)勢依賴于實(shí)驗(yàn)的生長條件.富Mo(Mo-rich)環(huán)境中Mo的化學(xué)勢為μMo=E(Mo),E(Mo)代表體心立方結(jié)構(gòu)的Mo金屬中單個Mo原子的能量.S的化學(xué)勢為μS=代表六方結(jié)構(gòu)的單層MoS2原胞的總能(下同);富S(S-rich)環(huán)境中S的化學(xué)勢為代表S2(g)的總能.Mo的化學(xué)勢為μMo=E(MoS2)?2μS.圖2給出了在富S和富Mo生長環(huán)境下單層MoS2中的點(diǎn)缺陷形成能,這里包括了空位(S和Mo)引起的缺陷和替位式摻雜(F,Cl,Br和I)引起的缺陷.從圖2中可以看出,由空位引起的缺陷,在富S環(huán)境下,Mo空位的缺陷形成能低于S空位的缺陷形成能;在富Mo環(huán)境下,S空位的缺陷形成能低于Mo空位的缺陷形成能.替位式摻雜情形下,富Mo生長環(huán)境的缺陷形成能小于富S生長環(huán)境的缺陷形成能,這說明對于替位式摻雜,富Mo生長環(huán)境更有利于摻雜.此外,S原子的替位式摻雜中,F,Cl,Br,I這四種元素相比,無論是富Mo還是富S環(huán)境,都表明F元素?fù)诫s的缺陷形成能最小,這說明F元素更容易摻入到單層MoS2.基于缺陷形成能的計(jì)算結(jié)果,下文中鹵族元素?fù)诫s的結(jié)構(gòu)模型都是考慮替代S原子位置.
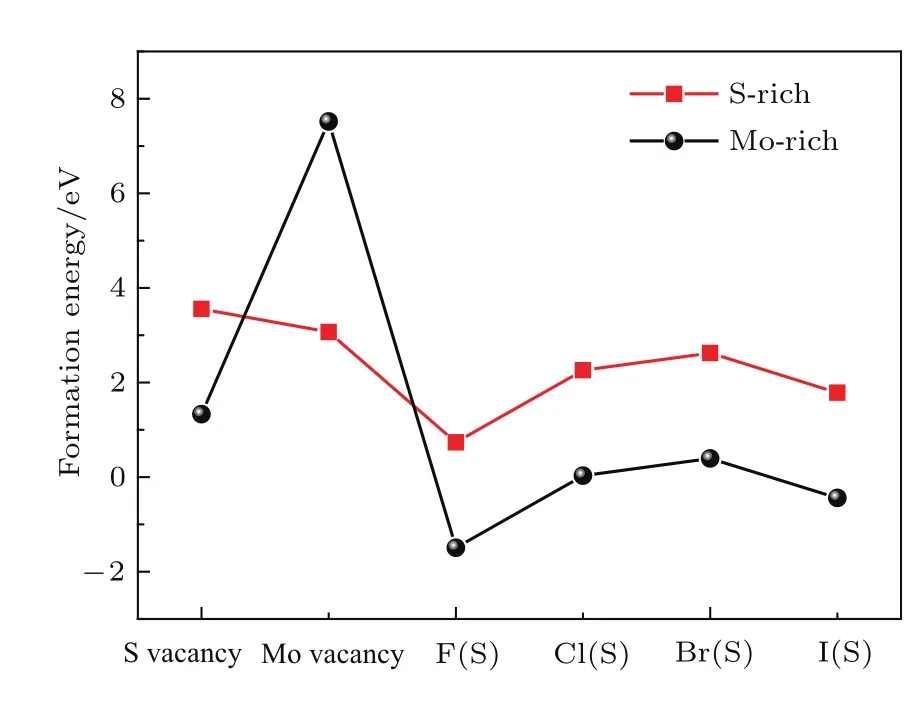
圖2 不同生長環(huán)境下MoS2中的點(diǎn)缺陷形成能Fig.2.Formation energies for various defects in MoS2under di ff erent growth conditions.
3.2 能帶結(jié)構(gòu)
圖3給出了鹵族元素?fù)诫s單層MoS2的能帶結(jié)構(gòu),作為比較,同時也一并給出了純單層MoS2的能帶結(jié)構(gòu).計(jì)算結(jié)果顯示純單層MoS2是直接帶隙半導(dǎo)體,其帶隙值為1.65 eV,如圖3(a)所示,這與其他的理論計(jì)算結(jié)果一致[24].鹵族元素?fù)诫s后的單層MoS2仍然表現(xiàn)為直接帶隙的半導(dǎo)體,且摻雜后的體系帶隙有所增大.所有鹵族元素的摻雜,都在帶隙中引入了雜質(zhì)能級,見圖3(b)—圖3(e)中紅色曲線.同時,費(fèi)米能級均發(fā)生了向?qū)У追较虻囊苿?并且費(fèi)米能級穿過了雜質(zhì)能級,說明鹵族元素?fù)诫s屬于n型摻雜.另外,計(jì)算結(jié)果顯示隨著摻雜原子半徑的增大,雜質(zhì)能級越來越平坦,相應(yīng)地雜質(zhì)能級上Γ點(diǎn)附近電子的有效質(zhì)量會變大.
3.3 肖特基勢壘高度
在金屬和半導(dǎo)體的n型接觸下,對于電子而言,其肖特基勢壘的高度Φe定義為
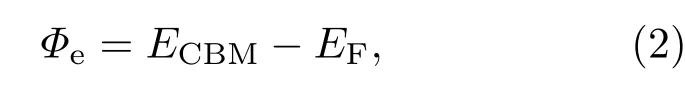
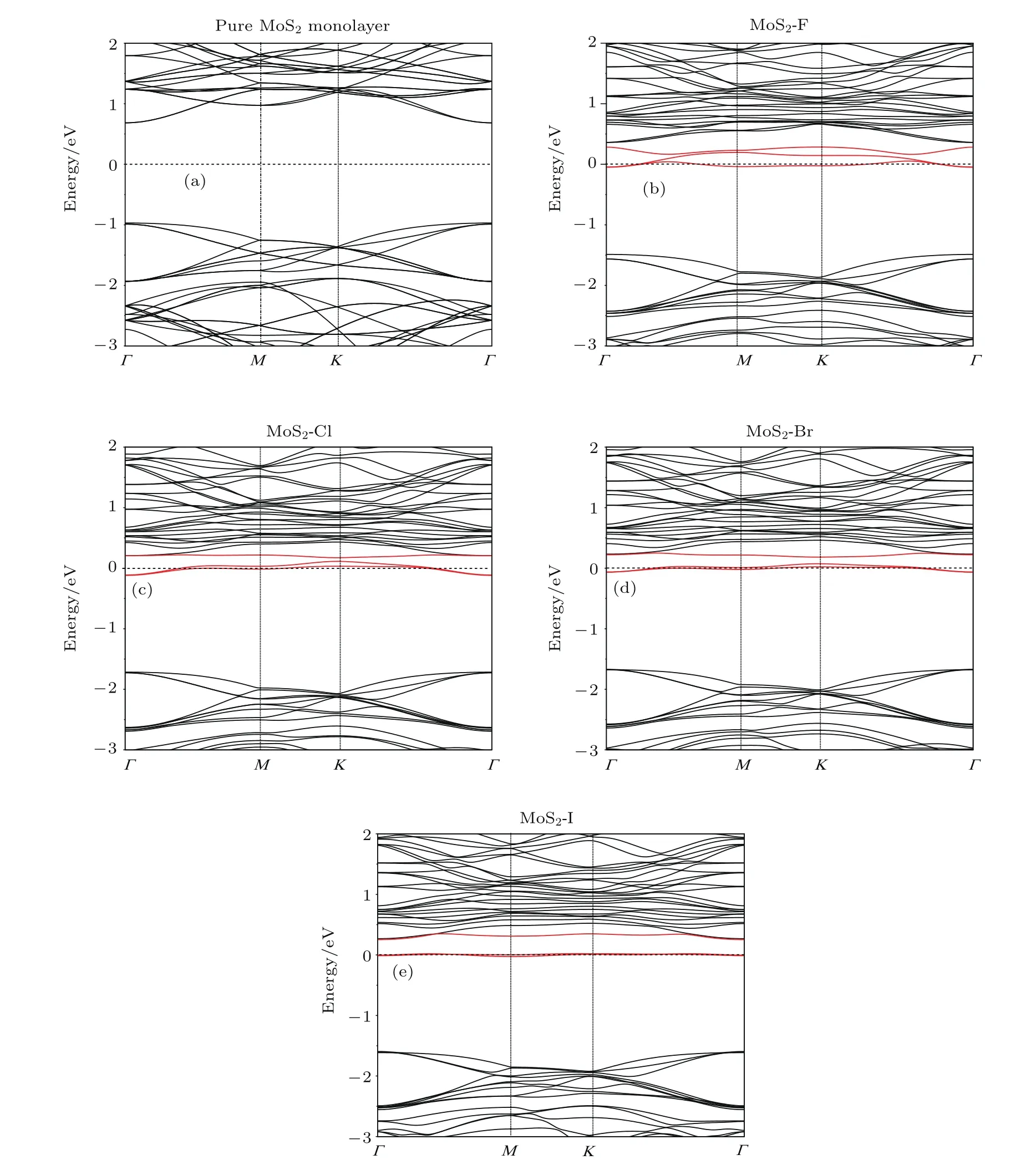
圖3 (網(wǎng)刊彩色)(a)單層MoS2能帶結(jié)構(gòu);(b),(c),(d)和(e)分別是F,Cl,Br和I摻雜單層MoS2后的能帶結(jié)構(gòu)Fig.3.(color online)(a)Band structure of the monolayer MoS2;(b),(c),(d),(e)band structures of the F,Cl,Br,and I-doped monolayer MoS2,respectively.
這里ECBM是指金屬-半導(dǎo)體界面體系中半導(dǎo)體能帶的導(dǎo)帶底能量,EF是體系的費(fèi)米能級.我們計(jì)算了Au-MoS2界面體系的能帶結(jié)構(gòu),如圖4(a)所示,這里紅色曲線為單層MoS2的能帶,灰色曲線為Au-MoS2的能帶,考慮金屬-半導(dǎo)體接觸對半導(dǎo)體原子內(nèi)層電子的能級幾乎沒有影響,同時考慮到靠近核的電子處于深層較低的能級,我們將單層MoS2和Au-MoS2的能帶的最低能級對齊得到了圖4(a),并由此得到了肖特基勢壘高度,鹵族元素?fù)诫s后肖特基勢壘高度也由類似的辦法得到.相比于純單層MoS2的能帶結(jié)構(gòu),Au-MoS2界面體系的費(fèi)米能級向?qū)У追较蛞苿?這表明單層MoS2與Au的接觸屬于n型接觸.根據(jù)(2)式,我們得到Au和單層MoS2之間的肖特基勢壘為0.69 eV.最近美國普渡大學(xué)葉培德教授課題組[21]從實(shí)驗(yàn)上比較了有無Cl氣體條件下金屬與單層MoS2的接觸電阻的大小.實(shí)驗(yàn)結(jié)果顯示,在Cl氣氛圍下,接觸電阻顯著降低,并猜測這是由于肖特基勢壘降低而導(dǎo)致的.為了驗(yàn)證他們對實(shí)驗(yàn)現(xiàn)象的解釋,我們計(jì)算了Cl摻雜的Au-MoS2體系的能帶.計(jì)算結(jié)果表明Cl摻雜后的體系費(fèi)米能級又向?qū)Х较虬l(fā)生了移動,如圖4(b)所示.同樣地,根據(jù)(2)式,Cl摻雜后Au-MoS2界面的肖特基勢壘變?yōu)?.58 eV,相比未摻雜的結(jié)構(gòu),肖特基勢壘明顯減小了.我們的計(jì)算結(jié)果從原子尺度且定量地驗(yàn)證了上述實(shí)驗(yàn)上的猜測.另外,我們也給出了其他鹵族元素?fù)诫s對肖特基勢壘的影響,見圖4(c).F摻雜后體系的肖特基勢壘最低,Br和I摻雜后體系的肖特基勢壘反而增高.
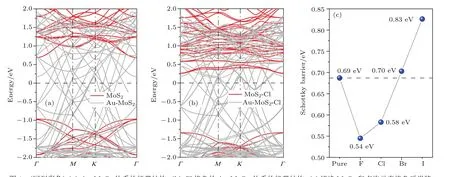
圖4 (網(wǎng)刊彩色)(a)Au-MoS2體系的能帶結(jié)構(gòu);(b)Cl摻雜的Au-MoS2體系的能帶結(jié)構(gòu);(c)純凈MoS2和鹵族元素?fù)诫s后肖特基勢壘高度Fig.4.(color online)(a)Band structure of the Au-MoS2system;(b)band structure of the Cl-doped Au-MoS2system;(c)Schottky barrier height of the pure MoS2and halogen-doped MoS2.
3.4 差分電荷密度和布居分布
直觀上看,Au-MoS2界面肖特基勢壘高度的改變主要源于體系費(fèi)米能級的變化,而費(fèi)米能級的移動主要是因?yàn)榻缑嫣庪姾赊D(zhuǎn)移以及化學(xué)鍵形成導(dǎo)致的.為了更詳細(xì)地分析不同鹵族元素?fù)诫s對Au-MoS2界面肖特基勢壘高度的調(diào)制,我們計(jì)算了鹵族元素?fù)诫s前后體系的差分電荷密度.差分電荷密度定義為
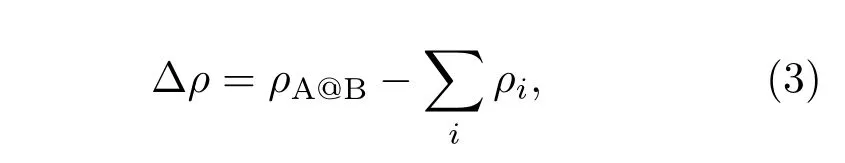
圖5 (網(wǎng)刊彩色)(a)Au-MoS2體系的差分電荷密度;(b)Cl摻雜Au-MoS2的差分電荷密度Fig.5.(color online)(a)The di ff erence of charge density for the Au-MoS2system;(b)the di ff erence charge density of the Cl-doped Au-MoS2.
式中ρA@B表示原子結(jié)合后體系的電荷密度;ρi表示孤立原子的電荷密度,下標(biāo)i遍及體系所有原子求和.該方法可以顯示由于原子鍵合引起的電子密度變化.?ρ的單位為electrons/?3.我們分別計(jì)算了Cl原子摻雜前后Au-MoS2體系的電荷密度差,并做了平行于z軸穿過摻雜原子的切片,得到了平面上的電荷密度分布,如圖5所示.圖5中藍(lán)色區(qū)域表示電子缺失的區(qū)域,紅色區(qū)域表示電子富集的區(qū)域.結(jié)果顯示,在Au-MoS2界面處,由于電荷轉(zhuǎn)移而形成了偶極矩.在Cl摻雜后,Au原子層和S_con(見圖1(e))原子層之間的電荷的轉(zhuǎn)移明顯減小,導(dǎo)致界面處偶極矩變小,使得費(fèi)米能級的發(fā)生向下移動,最終導(dǎo)致肖特基勢壘的高度被降低.
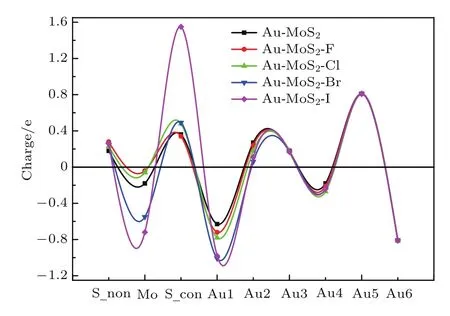
圖6 (網(wǎng)刊彩色)不同鹵族元素?fù)诫s前后體系的電荷分布Fig.6.(color online)The charge distributions of the di ff erent halogen-doped structure and pure structure.
此外,我們分別計(jì)算了不同鹵族元素?fù)诫s前后體系的電荷分布情況,通過計(jì)算布居分析,沿z軸對每一層原子的電荷進(jìn)行求和,做出了圖6.我們發(fā)現(xiàn)不同鹵族元素?fù)诫s下,每一層原子電荷量分布的趨勢是一致的.S_non(見圖1(e))層原子周圍集中了負(fù)電荷,Mo原子層上集中了正電荷,S_con層原子上集中了負(fù)電荷,Au1層原子上出現(xiàn)了大量的正電荷,這與經(jīng)典的金屬-半導(dǎo)體接觸理論所得結(jié)果是一致的,在金屬半導(dǎo)體界面區(qū)出現(xiàn)了電荷耗盡區(qū),金屬中的電子轉(zhuǎn)移到了半導(dǎo)體上.關(guān)于電荷轉(zhuǎn)移對界面處偶極效應(yīng)的貢獻(xiàn)可以利用平板電容模型來描述[25],即
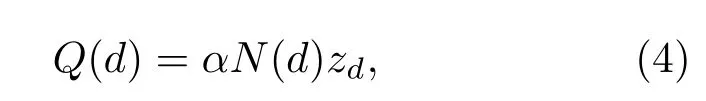
這里α=e2/(ε0A),A為單層MoS2單胞的面積;N(d)代表從每個MoS2單胞轉(zhuǎn)移到金屬上的電子數(shù);zd代表S_con和Au1之間轉(zhuǎn)移電荷層的有效距離.我們發(fā)現(xiàn)鹵族元素的引入對電荷層之間有效距離同樣具有不同的調(diào)制作用,這就導(dǎo)致zd的大小不同,從而影響了界面偶極矩的大小.
4 結(jié)論
本文通過基于密度泛函理論的第一性原理方法,研究了鹵族元素?fù)诫s對單層MoS2能帶以及對Au-MoS2界面肖特基勢壘高度的影響.研究結(jié)果顯示鹵族元素的摻雜使得單層MoS2呈現(xiàn)n型摻雜,且在帶隙引入雜質(zhì)能級.同時,F和Cl的摻雜可以有效地降低Au-MoS2接觸形成的肖特基勢壘高度,而Br和I的摻雜反而增大了肖特基勢壘的高度,其中Cl摻雜的研究結(jié)果很好地解釋了實(shí)驗(yàn)上在Cl氣氛下Au-MoS2接觸電阻顯著降低的物理機(jī)制.基于差分電荷密度,我們解釋了肖特基勢壘高度被調(diào)制的原因,是由于鹵族元素的摻雜導(dǎo)致了Au與單層MoS2界面處的電荷轉(zhuǎn)移形成的偶極矩發(fā)生了變化.不同元素?fù)诫s效果的差異,主要在于對界面處電荷轉(zhuǎn)移的貢獻(xiàn)大小不同.本文研究結(jié)果有助于更深入地理解二維材料與金屬界面的內(nèi)在物理機(jī)制,可以為后續(xù)的二維材料器件化設(shè)計(jì)與優(yōu)化提供必要的依據(jù).
[1]Novoselov K S,Geim A K,Morozov S V,Jiang D,Zhang Y,Dubonos S V,Grigorieva I V,Firsov A A 2004 Science 306 666
[2]Geim A K,Novoselov K S 2007 Nat.Mater.6 183
[3]Lee G H,Yu Y J,Lee C,Dean C,Shepard K L,Kim P,Hone J 2011 Appl.Phys.Lett.99 243114
[4]Yoon Y,Ganapathi K,Salahuddin S 2011 Nano Lett.11 3768
[5]Fang H,Chuang S,Chang T C,Takei K,Takahashi T,Javey A 2012 Nano Lett.12 3788
[6]Liu W,Kang J,Sarkar D,Khatami Y,Jena D,Banerjee K 2013 Nano Lett.13 1983
[7]Radisavljevic B,Radenovic A,Brivio J,Giacometti V,Kis A 2011 Nat.Nanotechnol.6 147
[8]Li L K,Yu Y J,Ye G J,Ge Q Q,Ou X D,Wu H,Feng D L,Chen X H,Zhang Y B 2014 Nat.Nanotechnol.9 372
[9]Gong K,Zhang L,Ji W,Guo H 2014 Phys.Rev.B 90 125441
[10]Wu M S,Xu B,Liu G,Ouyang C Y 2012 Acta Phys.Sin.61 227102(in Chinese)[吳木生,徐波,劉剛,歐陽楚英2012物理學(xué)報(bào)61 227102]
[11]Mak K F,Lee C,Hone J,Shan J,Heinz T F 2010 Phys.Rev.Lett.105 136805
[12]Splendiani A,Sun L,Zhang Y,Li T,Kim J,Chim C Y,Galli G,Wang F 2010 Nano Lett.10 1271
[13]Liu H,Neal A T,Ye P D 2012 ACS Nano 6 8563
[14]Popov I,Seifert G,Tománek D 2012 Phys.Rev.Lett.108 156802
[15]Zhang L Y,Fang L,Peng X Y 2015 Acta Phys.Sin.64 187101(in Chinese)[張理勇,方糧,彭向陽2015物理學(xué)報(bào)64 187101]
[16]Das S,Chen H Y,Penumatcha A V,Appenzeller J 2013 Nano Lett.13 100
[17]Liu W,Kang J,Cao W,Sarkar D,Khatami Y,Jena D,Banerjee K 2013 Proceedings of the IEEE International Electron Devices Meeting Washington,DC,USA,December 9–11,2013 p499
[18]Gan L Y,Zhao Y J,Huang D,Schwingenschl?gl U 2013 Phys.Rev.B.87 245307
[19]Liu D,Guo Y,Fang L,Robertson J 2013 Appl.Phys.Lett.103 183113
[20]McDonnell S,Addou R,Buie C,Wallace R M,Hinkle C L 2014 ACS Nano.8 2880
[21]Yang L M,Majumdar K,Liu H,Du Y C,Wu H,Hatzistergos M,Hung P Y,Tieckelmann R,Tsai W,Hobbs C,Ye P D 2014 Nano Lett.14 6275
[22]Segall M D,Lindan P J D,Probert M J,Pickard C J,Hasnip P J,Clark S J,Payne M C 2002 J.Phys.:Condens.Matter 14 2717
[23]van de Walle C G,Neugebauer J 2004 J.Appl.Phys.95 3851
[24]Cheng Y C,Zhu Z Y,Schwingenschl?gl U 2011 Phys.Rev.B 84 153402
[25]Khomyakov P A,Giovannetti G,Rusu P C,Brocks G,van den Brink J,Kelly P J 2009 Phys.Rev.B 79 195425
PACS:82.65.+r,73.20.–r,74.62.Dh,71.15.MbDOI:10.7498/aps.66.118201
First principles investigation of the tuning in metal-MoS2interface induced by doping?
Tao Peng-Cheng1)2)Huang Yan1)Zhou Xiao-Hao1)?Chen Xiao-Shuang1)Lu Wei1)
1)(National Laboratory of Infrared Physics,Shanghai Institute of Technical Physics,Chinese Academy of Sciences,
Shanghai 200083,China)
2)(University of Chinese Academy of Sciences,Beijing 100049,China)
27 December 2016;revised manuscript
1 March 2017)
Two-dimensional(2D)materials have shown great potential for electronic and optoelectronic applications.Among the 2D materials,molybdenum disul fi de(MoS2)has received great attention in the transition metal dichalcogenides family.Unlike graphene,2D MoS2can exhibit semiconducting properties and its band gap is tunable with thickness.A demonstration of a single-layer MoS2based fi eld-e ff ect transistor(FET)with a high on/o ffcurrent ratio(about 108)has aroused the considerable interest.Although 2D MoS2exhibits fascinating intrinsic properties for electronics,the contact may limit the device performance severely.In a real device such as FET,semiconducting 2D MoS2needs contact with a metal electrode,and a Schottky barrier is always formed at the semiconductor-metal interface.The formation of low-resistance contact is a challenge,which is important for achieving high“on”current,large photoresponse and high-frequency operation.Therefore,understanding and tuning the interfaces formed between metals and 2D MoS2is critical to controlling the contact resistance.In this work,some e ff orts have been made to investigate the 2D MoS2-metal interface in order to reduce the Schottky barrier height.By using the fi rst-principles calculations based on density function theory,we investigate the e ff ects of halogen doping-on metal-MoS2interface,including the formation energy of defect,electronic structure,charge di ff erence,and population.All calculations are performed using the ultrasoft pseudopotential plane wave method implemented in the CASTEP code.We use the generalized gradient approximation for the exchange and correlation potential as proposed by Perdew-Burke-Ernzerhof.Firstly,we calculate the formation energy to fi nd the thermodynamically stable positions for the halogen elements located in 2D MoS2.It is shown that the halogen elements tend to occupy the S site of a MoS2monolayer.Meanwhile,for the MoS2monolayer,the halogen doping may introduce the defect level into the forbidden gap and make the Fermi level shift.For the metal-MoS2interface,halogen doping can modulate its Schottky barrier height e ff ectively in terms of Schottky-Mott model.This is because the Schottky barrier height at the metal-semiconductor interface depends on the di ff erence between the Fermi level and the band edge position of the semiconductor.At the metal-MoS2interface,the Fermi level is partially pinned as a result of the interface dipole formation and the production of the gap states.Therefore,using di ff erent metals with di ff erent work functions cannot modify the Schottky barrier height e ff ectively.Here we demonstrate that F and Cl doping can reduce the Schottky barrier height,while Br and I doping can increase it.According to the results of the di ff erential charge density analysis,we can ascribe the tuning of Schottky barrier height to the in fl uence of the dipole caused by the charge transfer among the interfaces.This study can explain the relevant experimental results very well and provide a potential route to achieving low-resistance contact in the future applications of 2D materials.
Schottky barrier,MoS2,doping,density functional theory
10.7498/aps.66.118201
?國家自然科學(xué)基金(批準(zhǔn)號:11334008,61290301)資助的課題.
?通信作者.E-mail:xhzhou@mail.sitp.ac.cn
?2017中國物理學(xué)會Chinese Physical Society
http://wulixb.iphy.ac.cn
*Project supported by the National Natural Science Foundation of China(Grant Nos.11334008,61290301).
?Corresponding author.E-mail:xhzhou@mail.sitp.ac.cn
猜你喜歡
新世紀(jì)智能(數(shù)學(xué)備考)(2020年11期)2021-01-04 00:38:16
當(dāng)代陜西(2020年13期)2020-08-24 08:22:02
中國外匯(2019年17期)2019-11-16 09:31:14
制造技術(shù)與機(jī)床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國材料進(jìn)展(2016年10期)2016-12-26 06:50:20
濰坊學(xué)院學(xué)報(bào)(2016年2期)2016-12-01 13:00:11
新聞傳播(2015年11期)2015-07-18 11:15:04
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
