基于ReaxFF力場的對硝基苯酚臭氧氧化分子動力學模擬
2017-11-01 17:29:18王子民謝勇冰李曉霞鳴曹宏斌
物理化學學報 2017年7期
關鍵詞:體系
王子民 鄭 默 謝勇冰 李曉霞,4,* 曾 鳴曹宏斌,* 郭 力,4
(1中國礦業大學(北京)化學與環境工程學院,北京100083;2中國科學院過程工程研究所復雜系統國家重點實驗室,北京100190;3中國科學院過程工程研究所環境技術與工程研究部,北京100190;4中國科學院大學化學與化工學院,北京100049)
基于ReaxFF力場的對硝基苯酚臭氧氧化分子動力學模擬
王子民1,2鄭 默2謝勇冰3李曉霞2,4,*曾 鳴1曹宏斌3,*郭 力2,4
(1中國礦業大學(北京)化學與環境工程學院,北京100083;2中國科學院過程工程研究所復雜系統國家重點實驗室,北京100190;3中國科學院過程工程研究所環境技術與工程研究部,北京100190;4中國科學院大學化學與化工學院,北京100049)
本文采用基于ReaxFF反應力場的分子動力學方法(ReaxFF MD),利用自主研發的國際首個基于GPU加速的ReaxFF MD程序系統GMD-Reax和獨特的化學反應分析工具VARxMD,探索臭氧氧化對硝基苯酚的反應機理。通過模擬考察了300 K恒溫條件下臭氧氧化水中對硝基苯酚的行為,獲得了酚結構開環、CO2生成、主要自由基(·OH、·O2、·O)及團簇型自由基的數量演變趨勢,并可定性描述六元環開環和CO2生成均遵循偽一級反應動力學規律。反應機理分析表明酚類分子在水溶液中被臭氧氧化的路徑主要經過攫氫、六元環開環、碳鏈的氧化分解三個階段,也揭示了自由基和團簇型自由基在臭氧降解對硝基苯酚時所發揮的重要作用。本工作是應用ReaxFF MD分子模擬方法對常溫水環境下臭氧降解酚類污染物反應機理研究的一個嘗試,可為深入認識該機理及相關的實驗、理論研究提供一定的參考。
對硝基苯酚;臭氧;反應分子動力學模擬;自由基行為;反應機理
1 引 言
煤焦廢水處理是水環境治理中的重大問題之一,一直備受關注1,2。焦化廢水主要來源于原煤高溫干餾、焦爐煤氣初冷和化工產品精制等工業過程,含大量難降解有機污染物,主要包括酚類及其衍生物、喹啉類化合物、苯、吡啶、萘類化合物等。這些污染物可長期存在于自然環境中,危害人類及其它生物的生命健康3,4。隨著人類工業的發展,焦化廢水的處理方法在不斷優化改進,從最早多段曝氣的活性污泥法5到應用廣泛的生物法6,7,逐步提高了廢水中有機物的去除效率8。但是,這些方法并不能保證水質的絕對安全9,生物法處理的出水中仍含有少量苯系物、雜環化合物等有毒有機物質,因此針對生物段出水的深度處理非常重要。臭氧氧化法作為深度處理廢水的一種有效方法,具有無二次污染、反應迅速、適用范圍廣、氧化徹底等優勢10,逐漸成為人們關注的熱點。目前,臭氧氧化法已成功應用于紡織、皮革、石油化工廠污水的處理11,并逐步應用于焦化廢水的深度處理。
臭氧氧化包括自由基間接氧化和臭氧分子直接氧化兩種路徑。間接氧化反應比直接氧化更為迅速,可氧化的污染物種類也更多11。盡管自由基對臭氧氧化過程非常重要10,12,但目前的實驗手段還難以對自由基進行連續原位檢測,加上焦化廢水中有毒有機污染物組成復雜,人們對臭氧降解有機物的化學反應機理的認識并不全面。Utsumi等12利用電子自旋共振技術(ESR)檢測臭氧在水環境中產生的活潑·OH的濃度,獲得·OH在臭氧間接氧化過程中起主導作用的結論。常溫下臭氧在水中溶解度低13,臭氧氧化過程產生的·OH 在水中存活時間極短14,因此,充分認識·OH自由基在臭氧氧化中的作用,提高其數量和反應活性對提高臭氧的氧化效率非常重要。將臭氧化與其它方法聯用,如 O3/H2O215、O3/H2O2/UV16、催化臭氧氧化17,18都可能提高·OH自由基的濃度,強化臭氧化作用。利用高效液相色譜(HPLC)、離子色譜(ion chromatograph)或氣質聯用儀(GC-MS)可檢測臭氧降解酚類污染物中產物的化學結構,并大致推斷出酚-醌-羧酸-二氧化碳降解歷程19,20。盡管如此,由于不穩定的降解產物出現時間極短,常規實驗手段難以捕捉瞬時出現的中間體結構,也難以連續檢測降解反應過程中相關小分子自由基的數量和演變趨勢,無法獲得詳細的臭氧降解有機污染物的反應機理。近幾年,分子模擬方法的快速發展為探索臭氧降解焦化廢水中有機污染物的反應機理提供了另一條途徑。
反應分子動力學方法(ReaxFF MD,Reactive Force Field Molecular Dynamics)是由 van Duin和Goddard等21共同發展的化學反應力場ReaxFF與分子動力學相結合的一種模擬方法。ReaxFF21基于鍵級描述化學體系中的鍵相互作用,利用 DFT擬合與優化原子與原子間的相互作用參數,并通過電負性平衡方法(EEM)22動態更新模擬體系中每個原子的電荷。與量子化學方法相比,ReaxFF MD無需預先定義化學反應路徑,并可應用于較大規模體系(> 103個原子)的模擬。
近年來,ReaxFF力場得到快速發展,針對不同元素的力場參數不斷被提出23,24,擴展了ReaxFF的應用范圍,包括燃燒25、熱解26,27、催化28等過程的反應機理研究。其中,Rahaman等29發展了可應用于水環境下有機化學反應的ReaxFF力場參數,并用于描述氨基酸的異構化過程,獲得了甘氨酸在水環境中從中性到兩性離子狀態的轉變機理,并發現其中的質子轉移反應可能是經由單一水分子完成。姜丹丹等30利用ReaxFF MD模擬了以苯并芘為代表的焦化廢水在超臨界水環境下降解過程中的結構變化,得到了三種主要的降解機理。張金利等31通過ReaxFF MD研究在超臨界水環境下降解印染廢水中的分散橙 25(Disperse Orange 25,DO25),發現溫度、分子比例(DO25/H2O/O2)及反應時間對降解過程均有影響,其中自由基 HO·、HO2·等可加速 DO25的降解。這些應用顯示了ReaxFF MD在應用于水環境下降解有機污染物的反應研究的潛力。
為了進一步提高ReaxFF MD的計算效率和模擬規模,鄭默等32開發了國際上首個基于圖形處理器(GPU)加速的ReaxFF MD程序GMD-Reax。ReaxFF MD的GPU并行化使得大規模反應體系(~10000個原子)在桌面計算機上的快速模擬成為可能。劉健33和韓君易34等開發的 VARxMD(Visualization and Analysis of ReaxFF Molecular Dynamics)是國際上首個實現對 ReaxFF MD模擬軌跡蘊含的化學反應進行自動化系統分析和可視化的程序系統。VARxMD可識別兩個時刻之間的化學反應,自動獲得模擬過程中發生的完整化學反應列表,進而對物種進行分析統計,顯示物種和化學反應的 2D、3D結構,并基于全局物種結構特征篩選化學反應,為復雜化學反應的分析提供了便利。GMD-Reax與VARxMD的成功研發及應用25?27,35,顯著提高了模擬效率,減少了分析ReaxFF MD模擬軌跡所消耗的人力和時間,有助于獲得完整的化學反應機理信息。
本文采用基于 GPU 高性能計算的GMD-Reax,利用VARxMD獨特的化學反應分析能力,以對硝基苯酚作為焦化廢水中有機污染物的代表性組分36,37,對大規模有機污染物體系進行了臭氧氧化的ReaxFF MD模擬,從分子層面獲得了有機物降解規律以及降解過程中重要自由基的演化規律和影響,期望為進一步認識臭氧降解對硝基苯酚的機理提供幫助。
2 模擬策略
2.1 模型構建
利用 ReaxFF MD從分子水平探索臭氧氧化有機物的反應機理,構建合理的模型體系是模擬的基礎,大規模模型對描述反應多樣性至關重要,但是會受到后續模擬計算代價的制約。酚類污染物是焦化廢水中的特征有機污染物,生物法降解的效果不好38,本文選擇對硝基苯酚作為臭氧氧化的目標污染物。焦化廢水生化出水酚的實際濃度多為 100?300 mg·L?1,甚至更低39,40。從模擬的角度看,依照實際濃度構建的體系不利于觀察臭氧與對硝基苯酚的反應情況,也不利于得到降解過程中的規律。考慮到實際實驗中臭氧分子是連續通入,為了在可接受的計算代價內觀察到反應趨勢,本文假設酚分子附近局部反應環境中的臭氧過量。此外,體系中的水環境會帶來大量的分子間氫鍵,易形成水團簇結構,不僅消耗寶貴的計算資源,也使化學反應分析更為復雜。因此本文進一步假設酚類有機物周圍的反應環境為臭氧分子的數量多于水分子的數量。基于上述假設,同時為了考察羥基與臭氧的協同作用,構建了不同組成的臭氧氧化水中對硝基苯酚的模型體系,表1列出了體系的組成、性質及模擬溫度(為簡略表示,對硝基苯酚縮寫為p-NP)。
模型構建過程中,首先基于 Materials Studio(MS)41中的Modules模塊,使用Amorphous Cell中Construction功能構建臭氧-對硝基苯酚-羥基-水的初始結構模型,密度均設為 1.0 g·cm?3,各體系構建時均采用 Dreiding42力場。接著利用 MS中的Forcite模塊對初始體系進行幾何優化(Geometry Optimization),降低初始模型的能量,同時防止對硝基苯酚體系中各分子和重要官能團相互重疊。對優化后的初始體系在300 K下進行NVT(恒溫恒容)分子動力學馳豫,同樣采用Dreiding力場,模擬時長200 ps,模擬步長為1.0 fs,采用Berendsen43方法控溫。最后再次進行幾何構型最優化。圖1為含300個對硝基苯酚、500個臭氧、500個水分子的初始體系3D結構。
2.2 ReaxFF MD模擬細節及反應分析
ReaxFF力場通過鍵級來描述體系中的成鍵和斷鍵,ReaxFF與MD結合可模擬較大的體系,且不需要對反應路徑進行預先定義,有潛力研究臭氧降解酚分子的相關自由基和反應機理。本文的ReaxFF MD模擬研究采用NVT系綜,為了保持一致,模擬溫度采用與實驗溫度接近的300 K。模擬采用周期性邊界條件,Berendsen方法控溫,控溫系數為0.1 ps,同時采用velocity-Verlet算法動態地更新原子在每個時間步的坐標和速度,模擬步長為0.4 fs,共模擬200 ps。模擬程序采用基于GPU并行的GMD-Reax32,模擬在CentOS 5.4服務器(處理器型號為英特爾-至強E5620、頻率為2.4 GHz、內存為2 GB、GPU卡型號為C2050)上進行,并每隔0.4 ps輸出一次反應軌跡和鍵級軌跡。每兩幀軌跡對應的兩個時刻之間有大量化學反應發生;且由于該模擬體系含有大量水,在ReaxFF MD模擬體系中會出現大量的水團簇分子,因此模擬結果的反應機理分析是一項有挑戰性的工作。本文采用VARxMD程序系統33,34對ReaxFF MD模擬結果進行化學反應的自動分析和物種自動檢索,提升了化學反應機理探索的效率。
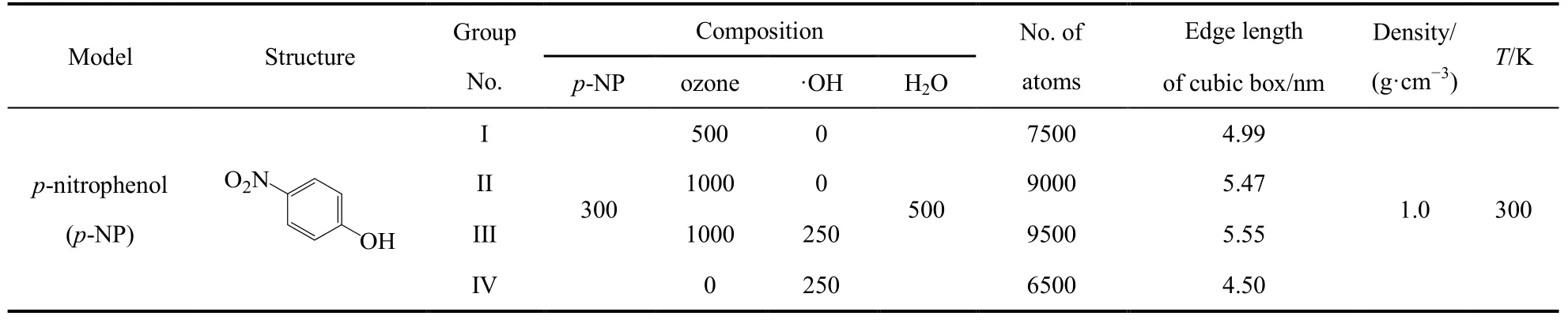
表1 利用Materials Studio構建的臭氧-有機物-水模型體系的組成和其它細節Table 1 Details of ozone-organic-water models constructed using Materials Studio.
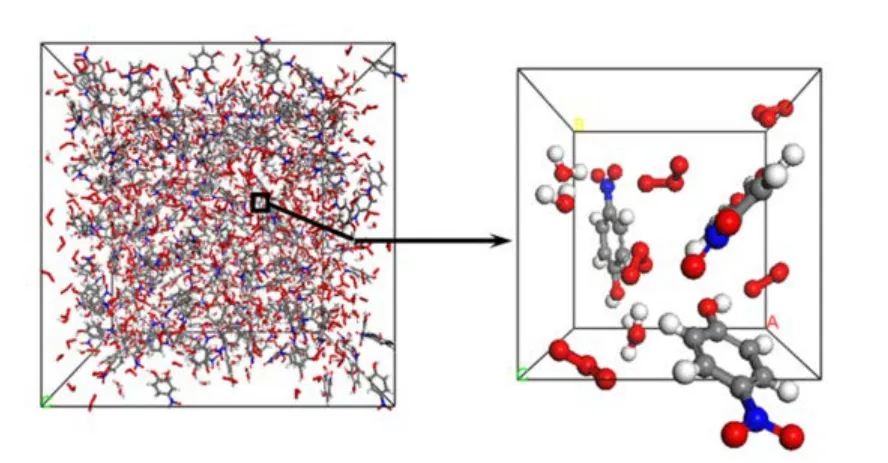
圖1 利用Materials Studio構建的含7500個原子的對硝基苯酚體系I(包括300個對硝基苯酚、500個臭氧、500個水)在300 K下幾何優化后的3D初始構象Fig.1 Optimized ozone-p-nitrophenol-water model containing 7500 atoms (model I, 300 p-nitrophenol,500 ozone and 500 H2O) at 300 K.
3 結果與討論
3.1 主要分子數量的演變趨勢
臭氧降解對硝基苯酚的終產物為水和二氧化碳,而芳香環開環是降解對硝基苯酚過程中的一個重要步驟,因此本文將芳香環和終產物 CO2的數量作為評價臭氧降解對硝基苯酚的兩個指標。在模擬開始的極短時間內臭氧與水快速反應生成大量自由基,這些自由基會促使酚環活化,使其形成攜帶自由基的芳香環(為簡略描述,下文統稱其為六元環)。圖2(a)中列出了采用ReaxFF MD模擬臭氧降解四個對硝基苯酚體系過程中六元環結構的數量隨時間的演化趨勢。對比不含羥基、不同臭氧含量的體系(I和II)發現,初始體系中臭氧數量的增加,可加快六元環開環速率。對比含1000個臭氧的兩個體系(II和 III)發現,在初始體系中額外加入一定數量的羥基自由基,也可加快六元環開環速率。為了考察開環速率,利用一級反應動力學對各體系六元環的數量演變趨勢進行擬合,其中含臭氧的體系相關系數均為0.98?0.99,遵循偽一級反應動力學規律。圖 2(b)是模擬四個體系過程中得到的 CO2數量隨時間的演變趨勢,可看出未加入臭氧、僅含 250個羥基自由基的體系(IV)自始至終均沒有生成CO2,而加入臭氧的三個體系在24?36 ps的階段開始生成CO2分子,其數量在36?200 ps的階段內隨時間穩步上升,且開環數量最多的體系中生成的 CO2分子數量也最多。利用一級反應動力學對此階段 CO2的數量演變趨勢進行擬合,得到相關系數為0.98?0.99。
通過圖2中六元環和CO2的數量演變可知,兩者數量均呈現較好的線性關系,說明利用ReaxFF MD模擬臭氧降解水中對硝基苯酚體系可定性描述其遵循偽一級反應動力學規律,該現象與楊德敏等44通過實驗得出的臭氧降解焦化廢水的生化出水化學需氧量(COD)符合表觀一級反應動力學的結論一致。
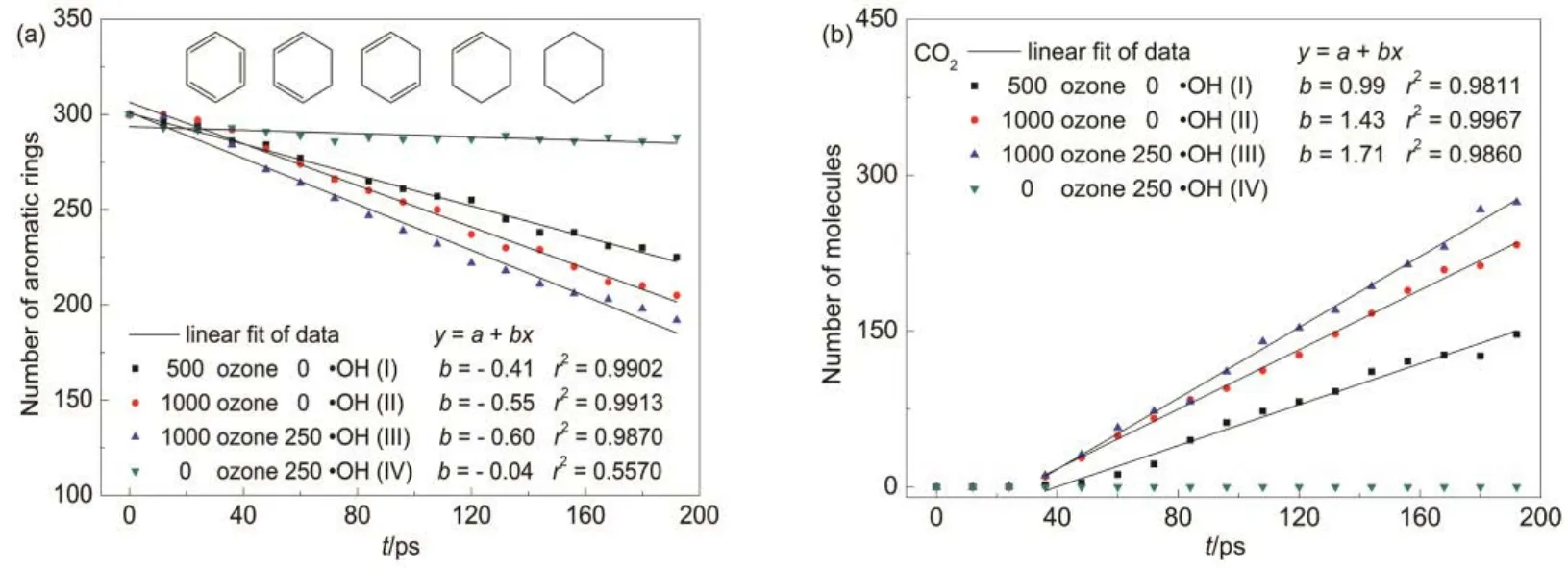
圖2 利用VARxMD得到的ReaxFF MD模擬臭氧降解水中對硝基苯酚(p-NP)體系在300 K條件下主要分子的數量隨時間的演變Fig.2 Evolution tendency of dominant molecules in ozonation of p-NP in water obtained from NVT ReaxFF MD simulation at 300 K by using VARxMD.(a) aromatic-ring; (b) CO2.
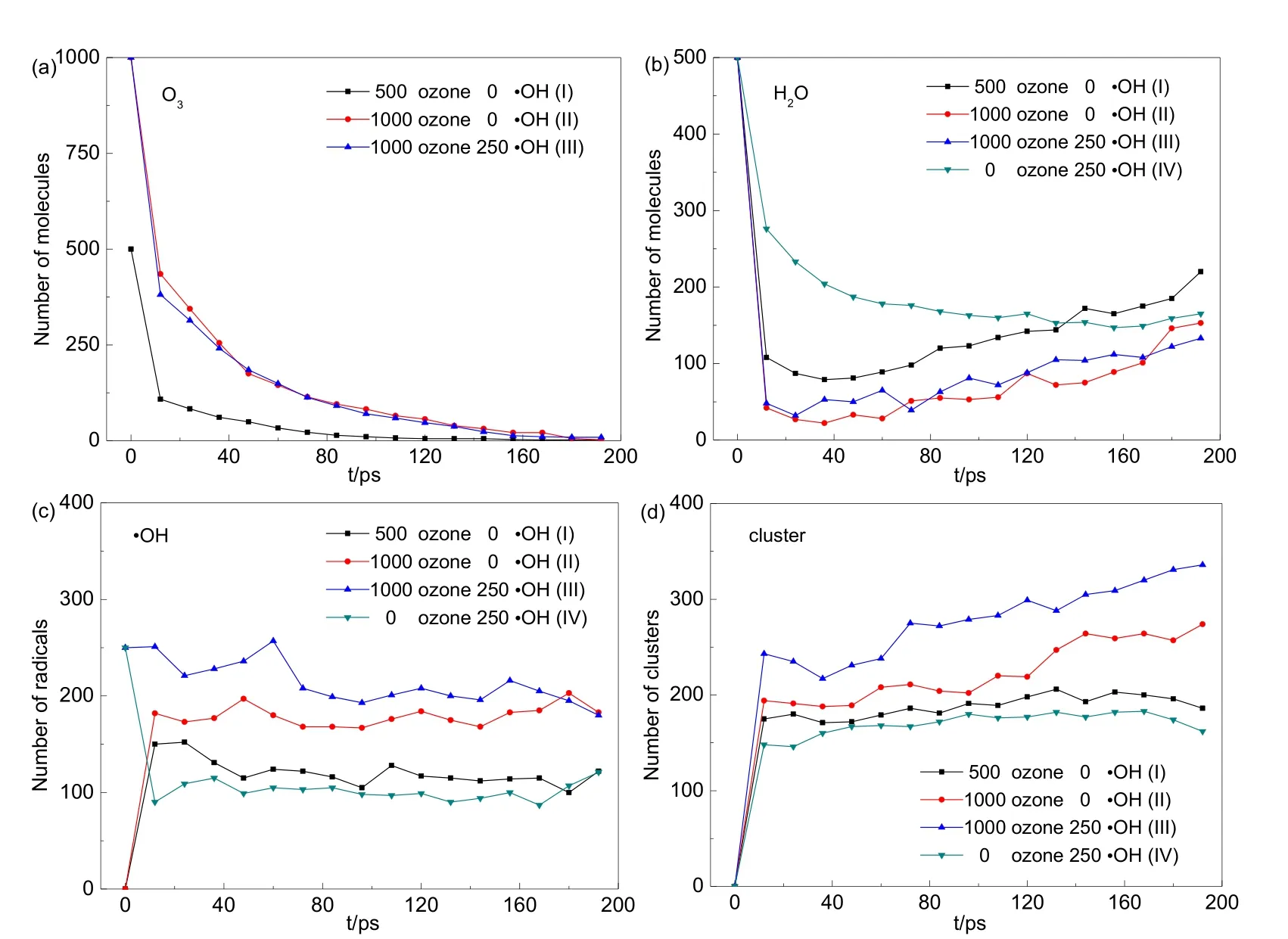
圖3 利用VARxMD得到的ReaxFF MD模擬臭氧降解水中對硝基苯酚(p-NP)體系在300 K條件下初始物質及團簇的數量隨時間的演變Fig.3 Evolution tendency of original components and clusters in ozonation of p-NP in water obtained from NVT ReaxFF MD simulation at 300 K by using VARxMD.(a) O3; (b) H2O; (c) ·OH; (d) cluster.
圖 3給出了模擬獲得的四個對硝基苯酚體系中初始物質臭氧、水、羥基及水團簇數量隨時間的演化趨勢。如圖3(a)所示,在僅含500個和1000個臭氧的體系(I和II)中,臭氧數量均在36 ps內快速下降了超過 70%,在 0?12 ps的下降速度最快;其后一直保持下降趨勢,雖然速度趨緩,但在200 ps時基本消耗殆盡。與此同時,如圖3(b)所示,體系I和II中的H2O的數量在0?12 ps階段快速下降至 108個(I)或 42個(含更多臭氧的體系II);其后,體系I、II中H2O的數量均在36 ps時下降到最低點(I中79個、II中22個),然后開始緩慢上升。圖 3(c)中,體系 I中的·OH數量在0?12 ps階段迅速上升至150個,體系II的·OH數量也上升至 182個的現象表明,臭氧的快速減少一部分生成了羥基自由基。圖 3(d)所給出的各體系中觀察到的團簇(包括 H2O-·OH、·OH-·OH 以及少量的H2O-H2O)數量隨時間演變趨勢表明,體系I的團簇數量在0?12 ps內快速上升至175個,體系II團簇數量也同時快速上升至194個,說明臭氧與大部分水快速反應,除了生成·OH自由基,一部分水可與·OH、或水與水分子結合成為團簇狀態,解釋了圖3 (b)中水快速下降的現象。當初始體系中臭氧數量增加,水分子數量下降幅度更大,所生成的游離·OH以及團簇數量增多。說明臭氧濃度的提升增加了體系中自由基及團簇的數量,可能以此促進對硝基苯酚的降解。
體系III因在含1000個臭氧的體系II中引入了250個·OH,其早期快速生成的·OH及團簇數量均比其他體系多。在不含臭氧的體系IV中,H2O及·OH的數量在模擬開始時均迅速下降,而團簇數量迅速上升,接著各物質數量在40?200 ps的階段內保持穩定。這一觀察說明,水體系中的 H2O及·OH均會相互結合形成團簇。此外,通過分析四個體系中水分子趨勢圖還可發現體系I、II、III中H2O數量在36?200 ps階段呈現緩慢上升狀態,而體系IV中H2O數量在此階段并未出現整體上升的現象。這是因為體系I、II、III在36 ps時已經有不同數量的對硝基苯酚分子被完全降解,生成CO2(圖2(b))和H2O,而體系IV中并未出現對硝基苯酚完全降解的現象。
通過各體系中臭氧、水分子、·OH及團簇數量的演變可知,臭氧與水分子反應會消耗大量水分子;此外,體系中不僅存在游離的·OH,還有大量·OH與水分子以團簇狀結構存在于水體系中。對硝基苯酚體系中六元環開環數量和·OH、團簇數量呈正相關關系,臭氧的存在可顯著加快開環速率以及 CO2生成速率,說明在構建體系時加入的臭氧在降解對硝基苯酚時起主要作用,·OH自由基和團簇可進一步提高中間產物降解為水和二氧化碳的效率。
3.2 臭氧降解對硝基苯酚中自由基的作用
臭氧降解對硝基苯酚時,臭氧遇水易分解生成大量自由基,因此自由基驅動的反應占主導45。通過 ReaxFF MD的模擬發現,絕大多數自由基為·OH、·O2、·O。考慮到體系可能呈現出多樣化的反應途徑、且有利于得到規律性結論,下文選取體系III,即含300個對硝基苯酚、1000個臭氧、250個羥基、500個水的體系作為考察重點。
3.2.1 小分子自由基
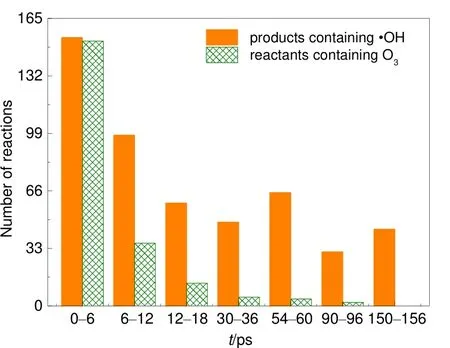
圖4 利用VARxMD得到的ReaxFF MD模擬臭氧降解水中對硝基苯酚體系(III)在300 K條件下與·OH有關的化學反應數量隨時間的演變Fig.4 Evolution trend of ·OH radical involved reactions in ozonation of p-nitrophenol in water obtained from NVT ReaxFF MD simulation at 300 K by using VARxMD (model III).
為了考察臭氧與·OH的協同作用,在構建p-NP體系III時除了1000個臭氧,額外加入了250個·OH自由基。模擬結果表明,該體系中六元環開環速率及 CO2生成速率均最快。并且從 3.1節的分析可知,臭氧與水反應可生成大量·OH自由基,體系III中·OH數量雖然多一些,但其演化并未發生太大變化(圖3(c)),在模擬過程中維持一個較穩定的狀態((220 ± 30)個)。為了解釋這種現象,對體系中·OH相關的化學反應演化進行了觀察,發現在不同的時刻均有大量·OH生成和消耗。為了大致得出·OH的生成情況,每隔6 ps提取一次反應列表,并對總反應列表中生成·OH的反應進行篩選,共得到2936個反應,其中由O3作為反應物參與的反應有264個。圖4給出不同時刻相關反應的數量,可看出在反應的起始時刻,即0?6 ps內幾乎所有·OH的生成反應都有O3參與;之后,由 O3參與生成·OH的化學反應迅速變少。由 O3參與生成·OH的化學反應主要集中在 0?54 ps階段內;在 54 ps后的階段內大部分·OH的生成與O3關系不大。借助VARxMD分析54 ps后的化學反應,發現體系中存在大量團簇型自由基,這些自由基主要由·OH、·O 等與水分子通過氫鍵結合形成,在所模擬的反應過程中,這些團簇型自由基也會發生內部斷鍵從而生成小分子自由基。該現象說明在反應起始階段,臭氧與水反應生成大量·OH,同時有幾乎同等數量的游離·OH參與形成團簇,這些團簇在之后模擬過程中會斷鍵形成新的游離·OH自由基,從而使體系中·OH數量維持在一個較穩定的范圍,見圖3(c)。
臭氧分解會生成大量的自由基,這些自由基在體系模擬中會相互轉化,如表 2所示。臭氧分解生成的·O2自由基演化如圖5(a)所示,在0?96 ps的階段內,體系中·O2自由基的數量生成趨勢與圖3(a)中臭氧消耗規律基本一致:在 0?12 ps內·O2的數量從0迅速升至572個,在12?96 ps內緩慢上升至815個;在96 ps后開始呈現出緩慢下降的趨勢。利用 VARxMD詳細觀察體系 p-NP中·O2的行為后發現:·O2主要參與 CO2的生成,通過與·CO反應生成·CO3自由基,接著O―O斷裂生成 CO2分子。如圖 5(a)所示,·CO3在 36 ps時首次出現,幾乎與圖 2(b)中 CO2分子在同一時間生成;而在 43?200 ps階段,·CO3自由基的數量起伏不定,這是因為·CO3自由基是CO2的主要前驅體,且·CO3→ CO2反應迅速,致使·CO3存在時間極短,因此各時間步出現的·CO3數量并沒有呈現出太強的規律性。

表2 利用VARxMD觀察ReaxFF MD模擬結果獲得的在300 K條件下臭氧降解水中對硝基苯酚過程中主要自由基的生成與消耗反應Table 2 Generation and consumption reactions of major radicals in ozonation of p-nitrophenol obtained by VARxMD in the 300 K-NVT ReaxFF MD simulations.
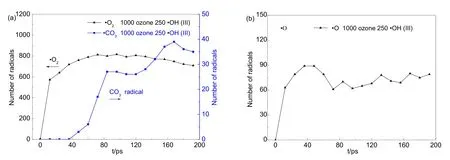
圖5 利用VARxMD得到的ReaxFF MD模擬臭氧降解水中對硝基苯酚體系(III)在300 K條件下主要自由基的數量隨時間的演變Fig.5 Evolution of dominant radicals in ozonation of p-nitrophenol in water (model III) obtained with aid of VARxMD in the 300 K-NVT ReaxFF MD simulation.(a) ·O2, CO3 radical; (b) ·O.
臭氧在水體系中除了生成·O2,還會生成少量的·O,同樣有助于其降解過程,如圖 5(b)所示。在前40 ps的模擬階段,體系p-NP中的·O的數量迅速上升,隨后·O的數量穩定為約 80個。借助VARxMD追蹤·O的消耗途徑,發現·O主要有四種消耗途徑:(1) ·O通過氫鍵作用與水分子團簇結合,使水團簇變為具有自由基性質的團簇,對六元環的開環有一定的幫助作用;(2) ·O相互結合,形成穩定的氧氣分子存在于密閉的模擬體系中;(3) ·O 之間相互結合形成具有進攻性的·O2自由基,一定程度上增加了臭氧直接分解的·O2數量,該反應途徑可能是·O2與·O 數量上有所差異的原因之一;(4) ·O 是生成·OH 自由基的一種中間體,·O與水分子結合形成·HOHO,接著斷鍵生成兩分子·OH自由基。
從上述分析可知,臭氧降解對硝基苯酚的過程與自由基·OH、·O2、·O的生成和消耗都有一定的關系,·OH與其它自由基或水分子形成團簇型自由基活化了整個體系,·O2主要作用于CO2的生成,·O通過上述四個途徑對幫助降解酚分子發揮一定的作用。
3.2.2 團簇型自由基
·OH的氧化能力強于臭氧46,在臭氧深度氧化酚類分子時起到至關重要的作用14,45。但是,通過本文對小分子自由基(·OH、·O等)的反應軌跡分析發現,在300 K恒溫條件下模擬臭氧降解對硝基苯酚體系中,·OH、·O 之間或者與水分子間更易以氫鍵相連,締結成大小不一的團簇型自由基,這些團簇型自由基的形成同樣需要消耗一定量的水分子,這應該是體系中水分子數量在模擬開始就迅速下降的一個原因(如圖3)。團簇型自由基具有和·OH、·O 等小分子自由基相似的反應活性,會攻擊酚分子的側鏈或六元環,發生加成反應或攫氫反應,從而促進酚的開環和降解。這一反應過程會加速六元環的開環和 CO2的生成,因此,團簇型自由基在各個體系數量的差異有可能是造成六元環的開環和 CO2的生成趨勢有所差別的原因之一(如圖2)。
為了進行區分,本文將含 7個及以上氫原子的HmOn團簇定義為大團簇,將含6個及以下氫原子的HmOn團簇定為小團簇,圖6是ReaxFF MD模擬得出的對硝基苯酚體系(p-NP)中大團簇和小團簇數量隨時間的演化趨勢。從圖6中可以看出,反應開始時的0?10 ps階段,體系p-NP中大團簇、小團簇數量均迅速升高,小團簇的數量更多,利用VRAxMD觀察可知,體系中最大團簇含H原子達到19個;在10?36 ps內大團簇數量繼續上升,而小團簇數量有少量下降。說明在模擬開始時,小分子自由基與水會隨機碰撞結合形成大小不一的自由基團簇,小團簇同樣會相互碰撞形成大團簇。在48?84 ps階段內大團簇數量出現大幅度波動,對應著O3參與生成·OH及其他自由基的化學反應的結束,說明大團簇的形成與O3密切相關。與此同時小團簇保持數量增多,猜測可能是O3生成的各類自由基促進大團簇斷鍵形成小團簇導致。84 ps之后大團簇數量緩慢下降,小團簇數量繼續緩慢上升。
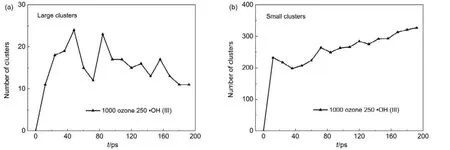
圖6 利用VARxMD得到的ReaxFF MD模擬臭氧降解水中對硝基苯酚體系(III)在300 K條件下的團簇分子數量隨時間的演變Fig.6 Evolution trend of water-clusters in p-nitrophenol model obtained from NVT ReaxFF MD simulation at 300 K by using VARxMD (model III).(a) large water-clusters (containing more than or equal to seven H atoms); (b) small water-clusters (containing less than or equal to six H atoms).
通過對比圖6中大、小團簇數量的演化猜測,在模擬過程中團簇的數量發生變化與對硝基苯酚的降解過程有一定的關系。利用VARxMD詳細觀察體系中團簇的行為,發現在臭氧降解酚的過程中,·OH直接進攻六元環使其開環的現象并不常見,更多的是具有自由基性質的團簇對六元環的進攻。這些團簇分子中大部分包裹·OH或·O自由基,·OH (·O)與水分子、·OH (·O)與·OH (·O)間通過氫鍵結合成大小不一的(·OH)x·(H2O)y形式的團簇分子,具有與自由基相似的攻擊性。與小團簇相比,大團簇所占的空間范圍更大,相對有更多的機會與酚分子發生碰撞。大團簇攻擊六元環使其支鏈增長形成大分子,在密閉盒子中可能會因空間構型的原因使大團簇支鏈斷裂成小團簇,因此會出現大團簇和小團簇數量交替上升或下降的現象。該模擬結果說明水體系中可能會存在團簇型自由基,更易進攻酚類污染物發生開環反應。
3.3 對硝基苯酚在ReaxFF MD模擬中降解機理分析
為了在可接受的模擬運行時間內讓大部分對硝基苯酚的降解反應發生,模擬中采用的時間步長為0.4 fs,一定程度上加速了氧化反應過程,也有可能掩蓋了一些反應的細節。借助VARxMD發現,臭氧降解對硝基苯酚的過程中,六元環開環后大多直接斷裂為小分子片段,并沒有經過多次開環閉環的振蕩反應。圖 7為模擬體系中含不同碳原子數的分子片斷的數量演化。
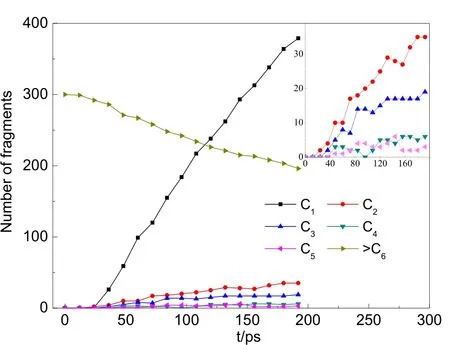
圖7 利用VARxMD得到的NVT ReaxFF MD模擬臭氧降解水中對硝基苯酚體系(III)在300 K條件下含C片斷數量(C1指片斷分子中含1個碳)隨時間的演變Fig.7 Evolution trend of carbon fragments in ozonation of p-nitrophenol in water (model III)obtained by VARxMD from the 300 K-NVT ReaxFF MD simulation (C1 represents fragment containing only one carbon).
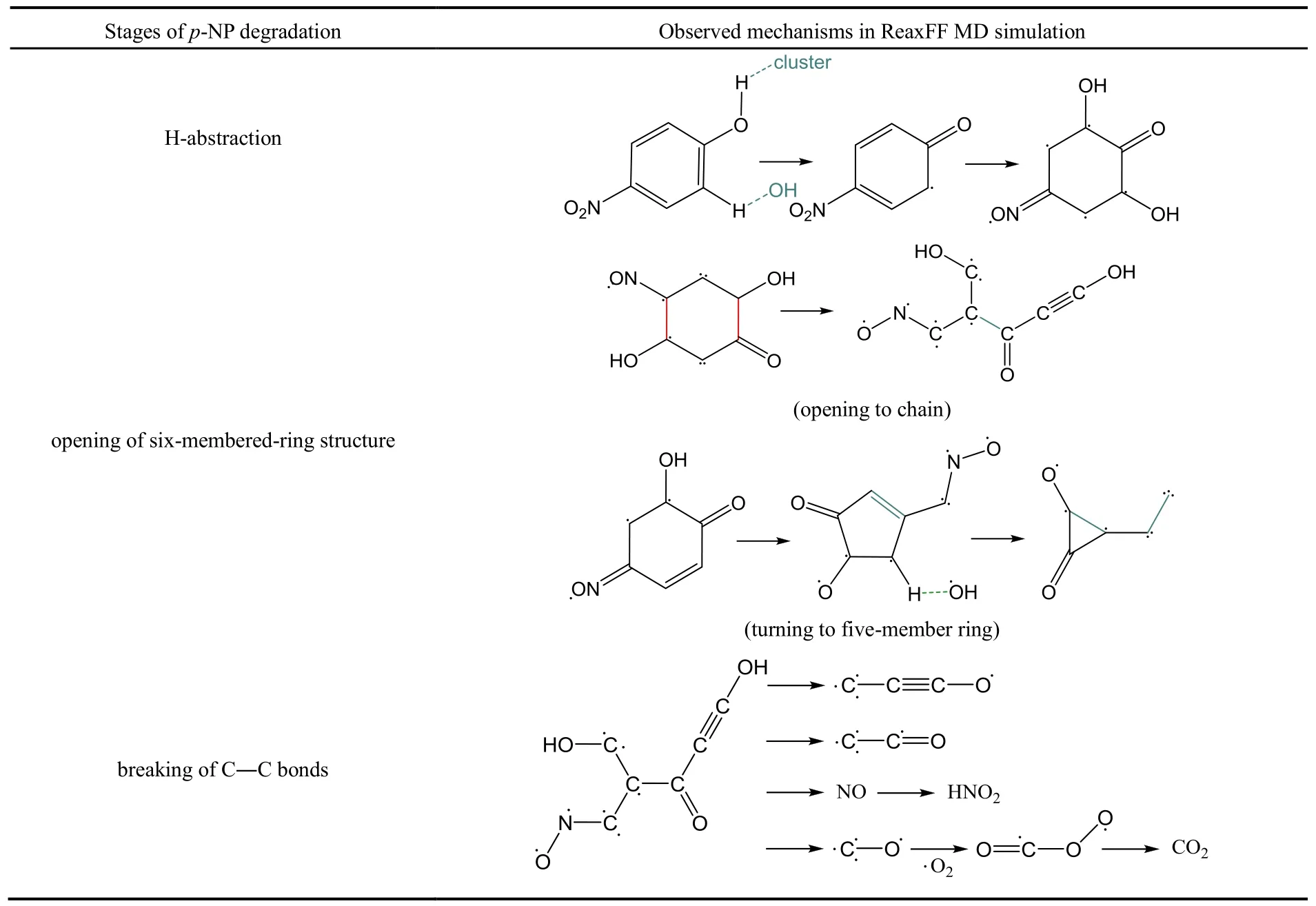
表3 通過VARxMD得到ReaxFF MD模擬臭氧降解對硝基苯酚體系在300 K-NVT的三個主要反應階段示例Table 3 Stages of p-nitrophenol degradation observed in 300 K-NVT ReaxFF MD simulation with the aid of VARxMD.
水環境中的酚分子間可能會因氫鍵作用與水分子等瞬時連結形成更大的分子團,由于未發生開環反應,圖7中將此與C6分子歸為一類,表示為“> C6”。如圖7所示,> C6物質數量隨著模擬時間的增加從300個緩慢減少至200左右,表示越來越多的六元環被臭氧降解為小分子片段。體系中 C1物質主要以·CO、·CO3、CO 及 CO2的形式存在,其分子數量從降解過程的30 ps開始迅速上升,生成速率遠快于其它的 C2?C5分子片斷。通過VARxMD分析發現,C2物質主要由六元環開環斷裂生成,它的數量同樣隨著時間緩慢上升。C2物質在降解過程中迅速轉化為·CO自由基和 CO2分子,使得 C1物質的數量快速上升。C4和 C5片段數量都比較少,僅有不足10個,生成趨勢不明顯。不同含碳片段數量的演化說明六元環開環形成鏈狀后更易于發生C?C均裂,形成C2、C3中間體,很少出現六元碳鏈中伯碳與仲碳斷鍵形成C1、C5中間體的現象。利用VARxMD可顯示兩個輸出時間步之間的反應物及生成物 2D、3D結構這一功能,可進一步跟蹤觀察到有機物在每一時間步的氧化情況,表 3給出對硝基苯酚在水環境下被臭氧降解過程的反應示例,可分為羥基攫氫、酚環開環、鏈狀結構斷裂分解三個階段:(1)酚環或者酚羥基上的攫氫反應,大多是由羥基或者含羥基的自由基團簇進攻酚環引發;自由基團簇中的氧與酚羥基中的氫形成氫鍵,導致酚羥基脫氫形成自由基;接著另一分子羥基或自由基團簇加成至羥基的α碳位,并繼續發生六元環的脫氫反應,形成苯醌結構。模擬觀察到的攫氫反應機理與通常認為的臭氧分子親電加成到苯環的機理有所差別,可能的原因是模擬時間步長較大(0.4 fs),缺失了部分的反應細節,也有可能是ReaxFF力場對該機理的描述不夠準確,需要進一步的工作加以證實。(2)羥基或自由基團簇對六元環的進攻,活化了六元環,其中,這些自由基更傾向于先進攻酚羥基的鄰位,這可由圖 8所示的開環過程中鄰位中間體分子數量遠大于間位中間體分子的觀察所佐證;當環上 C=O雙鍵或 C―O·單鍵自由基等結構越來越多,六元環的空間構型已發生很大形變,接著發生開環反應,大部分會形成鏈狀分子;小部分開環后會縮為五元環,經較長時間后轉化為小分子。(3)開環后的鏈狀分子含羰基、硝基等官能團,經較長時間的振蕩反應后斷鍵形成小分子結構。
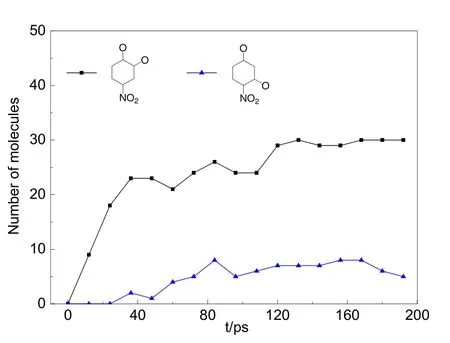
圖8 利用VARxMD得到的ReaxFF MD模擬臭氧降解水中對硝基苯酚體系(III)在300 K條件下酚羥基鄰、間位被進攻的中間體分子數量隨時間的演變Fig.8 Evolution trend of intermediates representing the p-nitrophenol’ reaction sites attacked by radicals in ozonation of p-nitrophenol in water obtained by VARxMD from the 300 K-NVT ReaxFF MD simulation of model III.
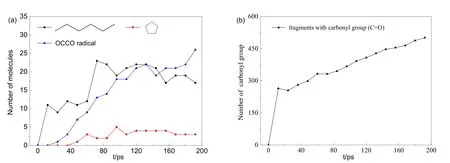
圖9 利用VARxMD得到ReaxFF MD模擬臭氧降解水中對硝基苯酚體系(III)在300 K-NVT降解條件下重要中間體片段隨時間的演變Fig.9 Evolution trend of dominant intermediates in the ozonation of p-nitrophenol in water obtained by using VARxMD from the 300 K-NVT ReaxFF MD simulation of model III.(a) key intermediates; (b) C=O moieties.
上述所觀察到的降解機理與圖 9中主要中間體的數量演化趨勢一致。如圖 9(a)所示,六元碳鏈的數量在0?70 ps處于上升階段,在70?150 ps處于動態穩定階段,在這一階段內既有鏈狀結構的生成也有碳鏈的分解,但須經過較長時間震蕩才會最終分解為小分子片斷,接著C6碳鏈分子有緩慢下降的趨勢;而五元環結構在40 ps時首次出現,其數量僅不足 5個,在隨后的模擬時間內處于穩定狀態。該現象說明大多數六元環開環后形成了碳鏈。C6碳鏈斷裂后形成的中間體多數轉化為 OCCO自由基,并分解為·CO,模擬體系中存在大量的·O2提高了·CO 與其碰撞的概率,加快了·CO 向 CO2轉變的過程。CO2的前驅體主要為·CO和含羧酸結構片段,并且·CO居多(體系III中發現僅有2個含羧酸結構片段)。據文獻報道在臭氧降解酚類分子的過程中會出現較多羧酸類中間體,但在模擬過程中并未發現太多此類中間體,這可能與體系中過多臭氧及羥基自由基有關。臭氧及羥基自由基的大量存在很容易使―COOH官能團中的氫或羥基脫落,形成H2O或·HO2、·HO3等自由基,導致―C=O結構數量增加,體系 III中―C=O結構(CO、CO2分子除外)數量演變如圖9(b)所示,其數量在0?12 ps階段迅速上升到264個,這是因酚羥基被體系中活性自由基攫氫生成酮所致;在24?200 ps階段緩慢上升,說明自由基進攻六元環后最終形成大量―C=O結構。
4 結 論
本文選取對硝基苯酚作為焦化廢水生物段出水的目標污染物,通過構建較大規模的模型,利用基于反應力場的ReaxFF MD模擬了對硝基苯酚在水環境下的降解反應。結合VARxMD分析工具,得到了體系中對硝基苯酚降解初始過程中重要自由基、中間體和產物隨時間的演變趨勢。
分析ReaxFF MD模擬結果的軌跡文件表明:在臭氧及羥基數量充足的水環境中,模擬可定性描述臭氧降解對硝基苯酚的初始反應行為,對硝基苯酚的降解與二氧化碳的生成遵循偽一級反應動力學規律。自由基的演化趨勢表明,O3在水環境中可快速分解形成·O2、·OH、·O等自由基,·O2在降解末期可加快中間產物向CO2轉化,提高降解速率,·O在對硝基苯酚的降解過程中以四種轉化路徑發揮其作用。此外,·OH、·O2、·O在水環境中易形成水-自由基的團簇,隱藏在團簇狀結構中的自由基使團簇有了攻擊性,可幫助六元環中C形成―C=O的結構,利于開環。借助VARxMD分析對硝基苯酚的降解路徑發現,酚類有機物在水環境下被臭氧降解的過程主要分為三步,攫氫、六元環開環、碳鏈的氧化分解。
值得注意的是,在水體系中可檢測出活性氧(·O2)的主要存在形式有兩種:單線態分子氧(1O2)和超氧自由基(·O2?),由于兩者電子排布不同使1O2與·O2?具有不同的化學活性,在臭氧催化氧化水中酚類有毒有機污染物時發揮了不同的作用47,48,由于模擬方法的限制,本文尚不能從·O2中嚴格區分出1O2和·O2?。
(1) Zhou, S. Y.; Watanabe, H.; Wei, C.; Wang, D. Z.; Zhou, J. T.;Tatarazako, N.; Masunaga, S.; Zhang, Y. Ecotox. Ecotoxicol.Environ. Safe. 2015, 115, 217. doi: 10.1016/j.ecoenv.2015.02.017
(2) Jin, X. W.; Li, E. C.; Lu, S. G.; Qiu, Z. F.; Sui, Q. J. Environ.Sci-China 2013, 25 (8), 1565. doi: 10.1016/S1001-0742(12)60212-5
(3) Zhu, Q.; Liu, Z. W.; Wang, J. R. Nonferrous Metal Science and Engineering 2011, 2 (5), 93. [朱 強, 劉祖文, 王建如. 有色金屬科學與工程, 2011, 2 (5), 93.] doi:10.13264/j.cnki.ysjskx.2011.05.003
(4) Wang, F.; Hu, Y. R.; Guo, C.; Huang, W.; Liu, C. Z. Bioresour.Technol. 2012, 110, 120. doi: 10.1016/j.biortech.2012.01.184
(5) Hall, D. A.; Nellist, G. R. J. Chem. Technol. Biotechnol. 1959, 9 (11),565. doi: 10.1002/jctb.5010091102
(6) Yang, P.; Wang, B. Environ. Protec. Chem. Ind. 2001, 21 (3), 144.[楊 平, 王 彬. 化工環保, 2001, 21 (3), 144.]doi: 10.3969/j.issn.1006-1878.2001.03.005
(7) Morper, M.; Jell, A. Linde Reports Sci. Technol. 2000, 62, 20.
(8) Han, T.; Chen, Z. S.; Lin, C.; Wen, Z. W.; Wei, C. H. Acta Sci.Circumst. 2016, 36 (1), 149. [韓 濤, 陳梓晟, 林 沖, 文澤偉,韋朝海. 環境科學學報, 2016, 36 (1), 149.]doi: 10.13671/j.hjkxxb.2015.0480
(9) Ren, Y.; Wei, C. H.; Wu, C. F.; Li, G. B. Acta Sci. Circumst. 2007,27 (7), 1094. [任 源, 韋朝海, 吳超飛, 李國保. 環境科學學報,2007, 27 (7), 1094.] doi: 10.13671/j.hjkxxb.2007.07.004
(10) Zhang, H.; Yang, Z. R.; Chen, H. Q. Environmental Protection 1995,No. 6, 11. [張 暉, 楊卓如, 陳煥欽. 環境保護, 1995, No. 6, 11.]doi: 10.14026/j.cnki.0253-9705.1995.06.004
(11) Chang, E. -E.; Hsing, H. J.; Chiang, P. C.; Chen, M. Y.; Shyng, J. Y.J. Hazard. Mater. 2008, 156 (1?3), 560.doi: 10.1016/j.jhazmat.2007.12.106
(12) Utsumi, H.; Hakoda, M.; Shimbara, S.; Nagaoka, H.; Chung, Y. S.;Hamada, A. Water Sci. Technol. 1994, 30 (9), 91.
(13) Wang, H. R.; Wang, S.; Li, Y. Q.; Wang, F. Y.; Li, Y. K.; Yin, J.Chinese Journal of Disinfection. 2009, 26 (5), 481. [王華然, 王 尚,李昀橋, 王福玉, 李迎凱, 尹 靜. 中國消毒學雜志, 2009, 26 (5),481.]
(14) Sun, H. F. The treatment of Organic Wastewater by Hydroxyl Radical Activated Oxygen. Ph. D. Dissertation, Beijing University of Chemical Technology, Beijing, 2010. [孫華峰. 羥基自由基活性氧處理有機廢水的研究[D]. 北京: 北京化工大學, 2010.]
(15) Roche, P.; Prados, M. Ozone Sci. Eng. 1995, 17 (6), 657.doi: 10.1080/01919512.1995.10555777
(16) Rosenfeldt, E. J.; Linden, K. G.; Canonica, S.; Gunten, V. U. Water Res. 2006, 40 (20), 3695. doi: 10.1016/j.watres.2006.09.008
(17) Wei, Q.; Qiao, S. F.; Sun, B. C.; Zou, H. K.; Chen, J. F.; Shao, L.RSC Adv. 2015, 5 (113), 93386. doi: 10.1039/C5RA14198B
(18) Tanaka, K.; Abe, K.; Hisanaga, T. J. Photochem. Photobiol. A 1996,101 (1), 85. doi: 10.1016/S1010-6030(96)04393-6
(19) Zazo, J. A.; Casas, J. A.; Mohedano, A. F.; Gilarranz, M. A.;Rodriguez, J. J. Environ. Sci. Technol. 2005, 39 (23), 9295. doi:10.1021/es050452h
(20) Valsania, M. C.; Fasano, F.; Richardson, S. D.; Vincenti, M. Water Res. 2012, 46 (8), 2795. doi: 10.1016/j.watres.2012.02.040
(21) van Duin, A. C. T.; Dasgupta, S.; Lorant, F.; Goddard, W. A. J. Phys.Chem. A 2001, 105 (41), 9396. doi: 10.1021/jp004368u
(22) Mortier, W. J.; Ghosh, S. K.; Shankar, S. J. Am. Chem. Soc. 1986,108 (15), 4315. doi: 10.1021/ja00275a013
(23) Mattsson, T. R.; Lane, J. M. D.; Cochrane, K. R.; Desjarlais, M. P.;Thompson, A. P.; Pierce, F.; Grest, G. S. Phys. Rev. 2010, 81 (5),054103. doi: 10.1103/PhysRevB.81.054103
(24) Chenoweth, K.; van Duin, A. C. T.; Goddard, W. A. J. Phys. Chem.A 2008, 112 (5), 1040. doi: 10.1021/jp709896w
(25) Liu, X. L.; Li, X. X.; Han, S.; Qiao, X. J.; Zhong, B. J.; Guo, L. Acta Phys. -Chim. Sin. 2016, 32 (6), 1424. [劉曉龍, 李曉霞, 韓 嵩, 喬顯杰, 鐘北京, 郭 力. 物理化學學報, 2016, 32 (6), 1424.]doi: 10.3866/PKU.WHXB201603233
(26) Zheng, M., Wang, Z., Li, X. X.; Qiao, X. J.; Song, W. L.; Guo, L.Fuel 2016, 177, 130. doi: 10.1016/j.fuel.2016.03.008
(27) Zhang, T. T.; Li, X. X.; Qiao, X. J.; Zheng, M.; Guo, L.; Song, W. L.;Lin, W. G. Energy and Fuel 2016, 30 (4), 3140.doi: 10.1021/acs.energyfuels.6b00247
(28) Castro-Marcano, F.; van Duin, A. C. T. Combustion and Flame 2013,160 (4), 766. doi: 10.1016/j.combustflame.2012.12.007
(29) Rahaman, O.; van Duin, A. C. T.; Goddard, W. A., III.; Doren, D. J.J. Phys. Chem. B 2011, 115 (2), 249. doi: 10.1021/jp108642r
(30) Jiang, D. D.; Zhang, J. L.; Wei, L.; Zhang, M.; Han, Y. Coking Wastewater Treatment via Supercritical Water: Reaxff Reactive Molecular Dynamics Simulation. In Engineering Science and Technology, Annual Conference of Chemical Industry and Engineering Society of China, Beijing, Oct 16–18, 2015; Chemical Industry and Engineering Society of China, Ed.; Chemical Industry and Engineering Society of China: Beijing, 2015; 414. [姜丹丹, 張金利, 李 韡, 張 銘, 韓 優. 基于ReaxFF力場對超臨界水處理焦化廢水反應機理的研究. In 工程科技, 2015年中國化工學會年會, 北京, Oct 16–18, 2015; 中國化工學會編; 北京: 中國化工學會, 2015: 414.]
(31) Zhang, J. L.; Gu, J. T.; Han, Y.; Li, W.; Gan, Z. X.; Gu, J. J. J. Mol.Model. 2015, 21 (3), 54. doi: 10.1007/s00894-015-2603-7
(32) Zheng, M.; Li, X. X.; Guo, L. J. Mol. Graph. Model. 2013, 41, 1.doi: 10.1016/j.jmgm.2013.02.001
(33) Liu, J.; Li, X. X.; Guo, L.; Zheng, M.; Han, J. Y.; Yuan, X. L.; Nie, F.G.; Liu, X. L. J. Mol. Graph. Model. 2014, 53 (9), 13.doi: 10.1016/j.jmgm.2014.07.002
(34) Han, J. Y.; Li, X. X.; Guo, L.; Zheng, M.; Qiao, X. J.; Liu, X. L.;Gao, M. J.; Zhang, T. T.; Han, S. Comp. & App. Chem. 2015, 32 (5),519. [韓君易, 李曉霞, 郭 力, 鄭 默, 喬顯杰, 劉曉龍, 高明杰,張婷婷, 韓 嵩. 計算機與應用化學, 2015, 32 (5), 519.]doi: 10.11719/com.app.chem20150502
(35) Li, X. X.; Zheng, M.; Han, J. Y.; Guo, L.; Liu, X. L.; Qiao, X. J.;Yuan, X. L. Scientia Sinica Chimica 2015, 45 (4), 373. [李曉霞, 鄭默, 韓君易, 郭 力, 劉曉龍, 喬顯杰, 袁小龍. 中國科學: 化學,2015, 45 (4), 373.] doi: 10.1360/N032014-00281
(36) Shao, N.; Wang, B. H.; Yuan, D. D. Contemp. Chem. Ind. 2015, 44(11), 2615. [邵 楠, 王寶輝, 苑丹丹. 當代化工, 2015, 44 (11),2615.] doi: 10.13840/j.cnki.cn21-1457/tq.2015.11.033
(37) Zhang, W. H.; Wei, C. H. Environmental Protection of Chemical Industry 2015, 35 (3), 273. [張萬輝, 韋朝海. 化工環保, 2015, 35(3), 273.] doi: 10.3969/j.issn.1006-1878.2015.03.010
(38) Liu, X. Q.; Li, G. B.; Wu, H. Z.; Zhang, W. H.; Guan, Q. Q.; Feng, C.H.; Wu, C. F.; Hu, Y.; Wei, C. H. Environ. Chem. 2012, 31 (10),1487. [劉顯清, 李國保, 吳海珍, 張萬輝, 關清卿, 馮春華, 吳超飛, 胡 蕓, 韋朝海. 環境化學, 2012, 31 (10), 1487.]
(39) Li, J. F.; Wu, J.; Sun, H. F.; Cheng, F. Q.; Liu, Y. Desalination 2016,380 (15), 43. doi: 10.1016/j.desal.2015.11.020
(40) Zhang, S. H.; Zheng, J.; Chen, Z. Q. Sep. Purif. Technol. 2014, 130(20), 610. doi: 10.1016/j.seppur.2014.06.019
(41) http://accelrys.com/products/collaborative-science/bioviamaterials-st udio (accessed Jan 18, 2017).
(42) Mayo, S. L.; Olafson, B. D.; Goddard, W. A. J. Phys. Chem. 1990,94 (26), 8897. doi: 10.1021/j100389a010
(43) Berendsen, H. J. C.; Postma, J. P. M.; van Gunsteren, W. F.; DiNola,A.; Haak, J. R. J. Chem. Phys. 1984, 81 (8), 3684. doi:http://dx.doi.org/10.1063/1.448118
(44) Yang, D. M.; Wang, B.; Yuan, J. M. Acta Petrolei Sinica (Petroleum Processing Section) 2012, 28 (4), 683. [楊德敏, 王 兵, 袁建梅.石油學報(石油化工), 2012, 28 (4), 683.] doi:10.3969/j.issn.1001-8719.2012.04.026
(45) Gharbani, P.; Khosravi, M.; Tabatabaii, S. M.; Zare, K.; Dastmalchi,S.; Mehrizad, A. Int. J. Environ. Sci. Tech. 2010, 7 (2), 377.doi: 10.1007/BF03326147
(46) Tang, Q. L. Ozonation of Various Organic Substances. Ph. D.Dissertation, Shanghai Jiao Tong University, Shanghai, 2013. [唐慶麗. 臭氧氧化降解多種有機污染物的研究[D]. 上海: 上海交通大學,2013.]
(47) Wang, Y. X.; Xie, Y. B.; Sun, H. Q.; Xiao, J. D.; Cao, H. B.; Wang,S. B. Catal. Sci. Technol. 2016, 6 (9), 2918.doi: 10.1039/C5CY01967B
(48) Wang, Y. X.; Xie, Y. B.; Sun, H. Q.; Xiao, J. D.; Cao, H. B.;Wang, S. B. ACS Appl. Mater. Interfaces 2016, 8 (15), 9710.doi: 10.1021/acsami.6b011
Molecular Dynamics Simulation of Ozonation of p-Nitrophenol at Room Temperature with ReaxFF Force Field
WANG Zi-Min1,2ZHENG Mo2XIE Yong-Bing3LI Xiao-Xia2,4,*ZENG Ming1CAO Hong-Bin3,*GUO Li2,4
(1School of Chemical and Environmental Engineering, China University of Mining and Technology, Beijing, Beijing 100083, P. R.China;2State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Science, Beijing 100190, P. R. China;3Research Center for Process Pollution Control, National Engineering Laboratory for Hydrometallurgical Cleaner Production Technology, Institute of Process Engineering, Chinese Academy of Science, Beijing 100190,P. R. China;4School of Chemistry and Chemical Engineering, University of Chinese Academy of Science,Beijing 100049, P. R. China)
Understanding the reaction mechanism of phenol ozonation in coking wastewater is very important for industrial applications of the ozonation process. Ozonation of p-nitrophenol in water at 300 K was simulated by ReaxFF force field molecular dynamics (ReaxFF MD) employing the GPU-enabled high-performance code of GMD-Reax and a unique code of VARxMD developed in authors’ group.Evolution trends of aromatic ring opening, CO2generation, dominant radicals (·OH, ·O2, ·O), and H2O clusters were obtained. The simulated CO2generation and reduction of aromatic ring could be described with pseudo-first-order kinetics. Moreover, the reaction pathways for the ozonation of p-nitrophenol can bedivided into three stages: hydrogen abstraction, opening of the six-membered-ring structure, and the breaking of C?C bonds. The simulations revealed the important role of radicals and water clusters in the ozonation of p-nitrophenol. This work is an attempt to investigate the ozonation mechanism of phenols in aqueous solutions at room temperature using ReaxFF MD, which should be helpful for further experimental or theoretical investigation of the mechanism.
p-Nitrophenol; Ozonation; ReaxFF MD; Radical behavior; Reaction mechanism
January 26, 2017; Revised: April 6, 2017; Published online: April 13, 2017.
O643
10.3866/PKU.WHXB201704132 www.whxb.pku.edu.cn
*Corresponding authors. LI Xiao-Xia, Email: xxia@ipe.ac.cn; Tel: +86-10-82544944. CAO Hong-Bin, Email: hbcao@ipe.ac.cn; Tel: +86-10-82544844.
The project was supported by the National Natural Science Foundation of China (21373227), the National Science Fund for Distinguished Young Scholars,China (51425405), and China’s State Key Laboratory of Multiphase Complex Systems, China (COM2015A004).
國家自然科學基金(21373227), 國家杰出青年科學基金(51425405)和多相復雜系統國家重點實驗室基金(COM2015A004)資助項目
? Editorial office of Acta Physico-Chimica Sinica
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
