TiO2納米管電極上電化學還原CO2生成CH3OH
2017-11-01 17:29:18裘建平童怡雯趙德明何志橋陳建孟
物理化學學報 2017年7期
裘建平 童怡雯 趙德明 何志橋 陳建孟 宋 爽,*
(1浙江工業(yè)大學環(huán)境學院,杭州 310032;2金華職業(yè)技術學院,浙江 金華 321007;3浙江工業(yè)大學化學工程學院,杭州 310032)
TiO2納米管電極上電化學還原CO2生成CH3OH
裘建平1,2童怡雯1趙德明3何志橋1陳建孟1宋 爽1,*
(1浙江工業(yè)大學環(huán)境學院,杭州 310032;2金華職業(yè)技術學院,浙江 金華 321007;3浙江工業(yè)大學化學工程學院,杭州 310032)
采用原位陽極氧化-煅燒法制備TiO2納米管(TiO2NTs)電極,運用X射線衍射(XRD)、電場發(fā)射掃描電子顯微鏡(FESEM)、X射線光電子能譜(XPS)、雙電位階躍測試等對制備電極進行表征,考察了其在0.1 mol·L?1KHCO3水溶液中電化學還原CO2的催化活性。結果表明TiO2NTs電極上電化學還原CO2的主產物為CH3OH,CH3OH由HCOOH和HCHO進一步還原而來。電極制備的最佳煅燒溫度為450 °C(TiO2NTs-450),電解電位?0.56 V (vs RHE (可逆氫電極))時反應120 min后,生成CH3OH的法拉第效率和分電流密度分別為85.8%和0.2 mA·cm?2。與550和650 °C煅燒的電極相比,TiO2NTs-450電極具有更高的催化活性,歸因于電極表面更多的三價鈦活性位,有利于CO2吸附,從而對·CO2?起到穩(wěn)定的作用,速率控制步驟轉變?yōu)椤O2?的質子化反應。
TiO2納米管電極;電化學還原;CO2;甲醇;還原機理
1 引 言
化石燃料燃燒會產生大量 CO2氣體導致溫室效應,并引發(fā)不可再生能源的枯竭1。CO2可被視作豐富且廉價的碳源用于生產燃料和有機化學品,將其轉化為能源燃料可產生顯著的環(huán)境效益和經濟效益。
將CO2還原為有機組分的非生物轉化有輻射2、熱3、光4以及電化學5等方法。其中,電化學還原 CO2具有操作條件相對簡便、反應過程易于控制、轉化率較高、可以利用潔凈可再生能源以及系統(tǒng)緊湊靈活、模塊化、易于放大生產等優(yōu)點,更具備實用性和潛在的工業(yè)應用價值。目前電化學法還原 CO2的電極主要為多晶單金屬電極,按催化轉化產物可分為兩大類:選擇生成CO電極(如Au、Ag、Zn),選擇生成甲酸電極(如 Sn、In、Pb)。
甲醇(CH3OH)因運輸、儲存方便,能量利用率高而被廣泛應用6,7。“甲醇經濟”模式可再循環(huán)大氣中過多的二氧化碳,并且CH3OH可作為可再生燃料或用于合成幾乎所有目前源自化石產品的原料。在水溶液中將CO2電化學還原為CH3OH主產物的電極主要有GaAs電極8、金屬電極(Mo、Pd)9,10和金屬氧化物電極(CuO)11。其中Canfield和Frese等9首次在 pH 為 4.2的 0.2 mol·L?1Na2SO4電解液中于 p-GaAs、n-GaAs電極上將 CO2電化學還原為CH3OH,但其生成CH3OH獲得高法拉第效率所需電極電位為?1.2 ? ?1.4 V (vs SCE(飽和甘汞電極)),遠低于該條件下CO2還原生成CH3OH的標準電極電位?0.536 V (vs SCE)。Mo電極能使生成CH3OH的法拉第效率達到 100%以上,主要源于Mo 電極的自腐蝕。而向 0.5 mol·L?1NaClO4電解液中加入0.01 mol·L?1吡啶可使Pd電極上生成甲醇的法拉第效率提高至 30%10。CuO電極在 0.5 mol·L?1KHCO3電解液中生成 CH3OH 法拉第效率達到38%11。
最近,Zhang等12對比了 Pt/RuO2/TiO2納米管電極和 Pt/RuO2/TiO2納米顆粒電極在 0.5 mol·L?1NaHCO3電解液中電化學還原 CO2的活性,發(fā)現TiO2納米管基電極表現出較高的CH3OH生成法拉第效率(60.5%),然而,TiO2納米管電極的晶相組成對電化學還原 CO2的影響及其電化學還原CO2為CH3OH機理目前未見報道。本文采用陽極氧化法原位制備具有高催化活性的TiO2納米管電極,考察煅燒溫度對電極物理化學性質及電極催化還原CO2活性的影響,并探討了CO2電化學還原為CH3OH機理。
2 實驗部分
2.1 TiO2NTs電極制備
將 20 mm × 20 mm × 0.5 mm 的 Ti片(純度99.99%)依次置于丙酮(AR,上海凌峰)和無水乙醇(AR,上海凌峰)中超聲清洗10 min,然后在體積比為1 : 3的HF(AR,40%(w,質量分數),阿拉丁)和HNO3(AR,68%(w),阿拉丁)混合溶液中刻蝕5 min,蒸餾水洗凈并用 N2吹干,最后作為陽極置于乙酸(AR,上海凌峰)和 HF混合電解液中(乙酸與0.5% HF體積比為1 : 7),陰極為25 mm × 25 mm × 0.5 mm的Pt片。控制電壓20 V下電解40 min,在預設溫度下煅燒2 h得到TiO2NTs電極。在450、550、650 °C下煅燒得到的電極,分別記為TiO2NT-450、TiO2NTs- 550和TiO2NTs-650。
2.2 催化劑表征
催化劑的晶相結構分析采用 X射線衍射儀(XRD,X?Pert PRO,PANalytical,荷蘭)。分析條件:Cu靶Kα射線,衍射光束波長為λ = 0.15418 nm,室溫,工作電流和電壓分別為40 mA和45 kV,衍射角2θ的掃描范圍為10°?80°,掃描速率為 2 (°)·min?1,掃描步長 0.01°。
催化劑的微觀表面形貌采用場發(fā)射掃描電子顯微鏡(FESEM,S-4800,Hitachi,日本)表征。催化劑的元素成分、表面電子態(tài)等采用X射線光電子能譜(XPS,PHI-5000C ESCA,Perkin-Elmer,美國)表征。
電極粗糙因子的測定方法:在 0.5 mol·L?1NaClO4溶液中,控制溫度為20 °C,電極電位先設定為0.1 V (vs SCE),10 s后再階躍至0.095 V (vs SCE)保持10 s,其計算如公式(1)所示13。
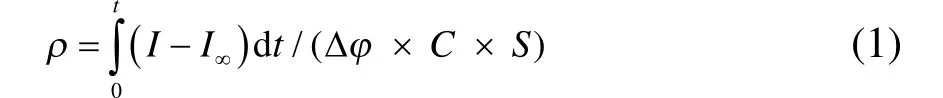
式中,ρ為粗糙因子(cm2·cm?2),I為反應電流(A),I∞為階躍后的穩(wěn)定電流(A),S為電極幾何面積(cm2),C 為理想雙電層間電容(20 μF·cm?2),t = 10 s,Δφ = 5 mV。
2.3 電化學還原CO2實驗
電化學還原CO2在自制的密閉H型反應池中進行。柱形陰極室直徑50 mm,高150 mm,有效容積295 mL,陰極室密封并設有取樣口及研究電極和參比電極接口;柱形陽極室的直徑 6 mm,高 150 mm,有效容積424 mL。陰陽極室之間用Nafion-117陽離子交換膜分隔。實驗中陰極液為100 mL CO2飽和的0.1 mol·L?1KHCO3水溶液,陽極液為180 mL 0.5 mol·L?1NaOH水溶液,反應溫度由低溫恒溫槽(THD-2015;寧波天恒儀器廠,中國)控制為25 °C。CO2還原反應由電化學工作站(CHI-660D,CH Instrument,美國)的時間-電流曲線功能控制進行恒電位電解,反應時間為120 min,在不同時間取樣測定產物的濃度。實驗中的電極電位通過公式(2)轉化為可逆氫電極電位。
E(vs RHE) = E(vs SCE) + 0.2412 V +0.0591 V × pH (2)
氣相產物 H2和 CO利用氣相色譜儀(7890B,Agilent,美國)分析檢測,配置HP-MOLESIEVE色譜柱(30 m × 320 μm × 12 μm),熱導檢測器(TCD)檢測。
氣相和液相中的 CH3OH均采用氣相色譜儀(6890N,Agilent,美國)檢測分析,配置了 30 m × 250μm × 25 μm的HP-INNOWAX毛細管柱,檢測器為FID。液相產物HCHO通過乙酰丙酮分光光度法進行測定14。
液相 HCOO?采用離子色譜儀(ICS 2000;Dionex,美國)測得,雙活塞泵(in series),IonPac AS19分析柱(4 mm × 250 mm;Dionex)分離,IonPac AS19保護柱(4 mm × 250 mm;Dionex),電導檢測器(DS6;Dionex),洗脫液使用陰離子電解抑制器(4 mm ASRS;Dionex)。
反應電極的催化選擇性、活性分別用產物的法拉第效率(FE)和分電流密度(PCD)來表征,FE可由公式(3)計算:
FE = znF/Q (3)其中,z為CO2還原為產物過程中的電子轉移個數,n為反應產物的摩爾數(mol),F為法拉第常數(96485 C·mol?1),Q 為反應過程中輸入的總電量(C)。
PCD可由公式(4)計算:
PCD = jtotal× FE (4)
其中,jtotal為反應的總電流密度。
3 結果與討論
3.1 催化劑的物理化學表征
3.1.1 XRD
不同溫度煅燒的TiO2NTs電極的XRD測定結果如圖1所示,由圖1(a)可知,所有樣品在2θ衍射角為 35.1°、38.4°、40.1°、53.0°和 70.7°出現衍射峰,分別對應于 Ti的(100)、(002)、(101)、(102)和(103)晶面(JCPDS卡片號05-0682),這主要源于Ti基底,由圖 1(a?),在 2θ 為 25.3°處均出現了銳鈦礦 TiO2(101)晶面衍射峰(JCPDS卡片號21-1272),而當煅燒溫度升至550 和650 °C時,在 27.4°出現了金紅石相 TiO2(110)晶面特征衍射峰(JCPDS卡片號 65-0190),且衍射峰隨煅燒溫度升高而增強,說明了450 °C煅燒獲得的電極為純銳鈦礦相,550和650 °C時得到的電極則為銳鈦礦和金紅石的混相。根據Spur-Myers公式15計算可得,TiO2NTs-550和TiO2NTs-650電極片中金紅石相的比例分別為0.64和0.92。
3.1.2 XPS
采用XPS分析不同溫度煅燒的TiO2NTs電極表面元素組成及元素化學價態(tài)。TiO2NTs-450、TiO2NTs-550、TiO2NTs-650電極的全譜圖和高分辨窄譜圖如圖 2所示,所有元素的結合能位置均根據C 1s峰(結合能284.6 eV)進行荷電校正。圖2(a)為所制備TiO2NTs電極的全譜圖,譜圖中出現了O、Ti、C的特征峰,表明所制備催化劑中有O、Ti、C三種元素存在。
TiO2NTs催化劑表面Ti 2p的高分辨XPS窄譜圖見圖2(b),一般而言,在Ti 2p窄譜圖中,結合能位置在457.4、458.5、463.3和464.4 eV的四個特征峰,分別對應于Ti3+2p3/2、Ti4+2p3/2、Ti3+2p1/2和 Ti4+2p1/2。用 Shirley法扣除本底,并用Voigt函數(Lorentzian-Gaussian卷積法)對Ti 2p圖進行分峰擬合16,具體結果如表 1所示,由表可知TiO2NTs電極上三價鈦(Ti(III))含量隨著煅燒溫度的升高而逐漸減少,TiO2NTs-450、TiO2NTs-550和TiO2NTs-650的Ti(III)原子百分含量分別為33.7%、16.6%和10.9%。
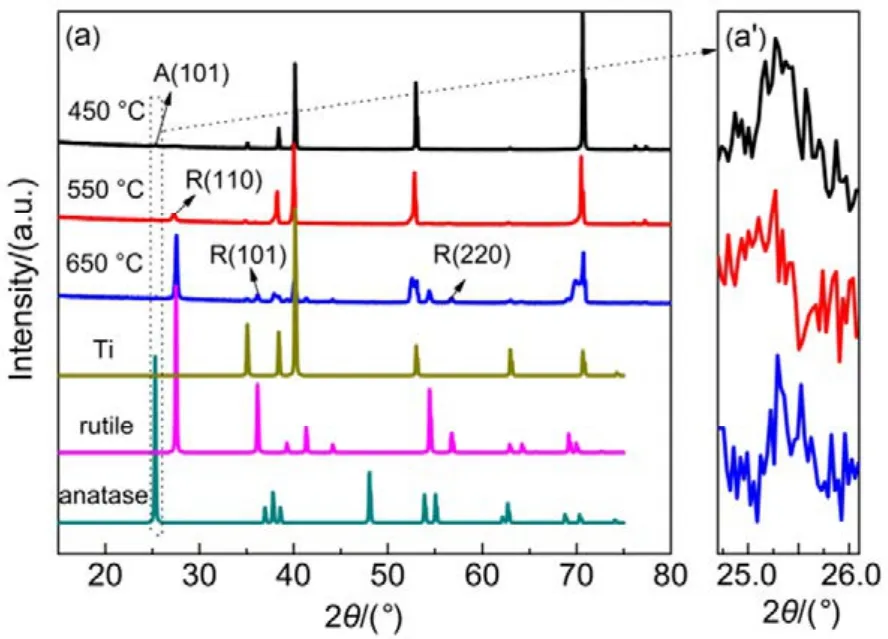
圖1 TiO2NTs-450、TiO2NTs-550和TiO2NTs-650的XRD圖譜Fig.1 XRD patterns of TiO2NTs-450,TiO2NTs-550 and TiO2NTs-650.
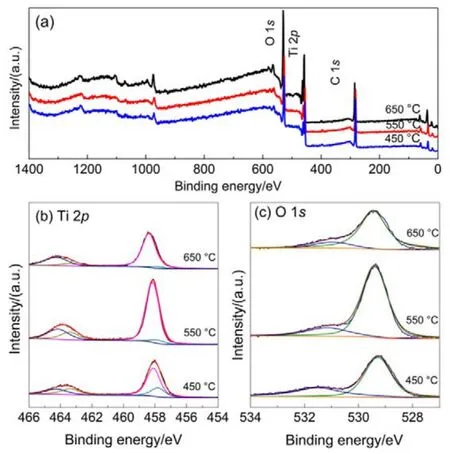
圖 2 TiO2NTs-450、TiO2NTs-550、TiO2NTs-650的XPS全譜圖(a)以及Ti 2p (b),O 1s (c)高分辨窄譜圖Fig.2 Wide-scan XPS spectra of (a) TiO2NTs-450,TiO2NTs-550 and TiO2NTs-650, as well as high-resolution scanning XPS spectra of(b) Ti 2p and (c) O 1s.
圖2(c)為各TiO2催化劑樣品的O 1s窄譜圖。如圖所示,O 1s峰去卷積分為3個特征峰:O 1s結合能在529.5 eV處的主要特征峰歸屬于晶格氧(Ti-O-Ti),而結合能在531.8 eV位置出現的特征峰為氧空位(O*),化學吸附的水分子在結合能532.8 eV位置處。由表 1可以看到,TiO2NTs- 450、TiO2NTs-550、TiO2NTs-650的表面氧空位占總含氧量的比例分別為33%、16.5%和13.6%,結果與Ti(III)變化一致。
3.1.3 FESEM
為研究煅燒溫度對TiO2NTs電極表觀形貌的影響,對其進行了FESEM測試。由圖3可知,所制備的TiO2納米管陣列有序緊湊,分布均勻,管徑范圍為69?82 nm,管壁厚度為24?27 nm。此外,通過對比納米管的形貌還可以看出,當煅燒溫度為450 °C時,TiO2NTs表面上TiO2納米管分布更為均勻,孔徑相對較大。當煅燒溫度高于550 °C時,部分納米管管口開始坍塌,并出現少量團聚現象。這是因為高溫煅燒時,TiO2發(fā)生銳鈦礦相向金紅石相轉變(從XRD圖中可證實),而這種晶相轉變導致TiO2晶格體積膨脹,且向外界釋放了大量的熱量,同時,銳鈦礦相向金紅石相的轉變引起 TiO2晶粒生長過快引發(fā)納米管坍塌現象,從而造成粒徑增大,電化學表面積減少17。
3.1.4 粗糙因子
為考察煅燒溫度對電極粗糙程度的影響,通過雙電位階躍法對 TiO2NTs-450、TiO2NTs-550和TiO2NTs-650電極的粗糙因子進行測定,根據式(1)可計算得到 TiO2NTs-450、TiO2NTs-550和TiO2NTs-650電極的粗糙因子分別為 875.3、723.7 和 540.3 cm2·cm?2,說明煅燒溫度升高會導致電化學表面積下降。
3.2 電催化還原CO2的活性和選擇性
為探明 TiO2NTs-450、TiO2NTs-550和TiO2NTs-650電極上電催化還原CO2活性,考察了反應時間為 120 min 時,電解電位(?0.16 ? ?1.16 V)對各產物FE和PCD的影響。從圖4中可以看出,受電極電位和檢測方法檢出限兩方面的影響,對于不同的電極,CO在電解電位負于?0.36 V時才能檢測到,HCHO則在電解電位負于?0.56 V時檢出,對應于 TiO2NTs-450、TiO2NTs-550和TiO2NTs-650電極,HCOOH則在電解電位分別負于?0.36、?0.46和?0.56 V時才能檢測到。CH4及其他二碳還原產物在研究的電極電位范圍內均未檢出。
在圖 4(a)、(b)和(d)中,三個電極上 FECO、FEHCOOH和 FECH3OH隨電解電位變化呈現相同趨勢,均隨電解電位負移先增加后下降,當電位為?0.56 V時達到最大值,此時TiO2NTs-450電極上FECO、FEHCOOH和 FECH3OH分別為 7.2%、4.5%和85.8%,TiO2NTs-550電極上 FECO、FEHCOOH和FECH3OH則為5.2%、2.5%和78.6%,TiO2NTs- 650電極上 FECO、FEHCOOH和 FECH3OH分別是2.7%、2.0%和72.2%,而FEH2則呈現相反的趨勢。對于TiO2NTs-450、TiO2NTs-550和TiO2NTs- 650電極,FEHCHO從?0.56 V的2.1%、1.05%和0.92%下降至?1.16 V的0.17%、0.10%和0.09% (圖4(c))。CO2的還原產物中FECH3OH最高,在電解電位為?0.56 V時,對應于 TiO2NTs-450、TiO2NTs-550和TiO2NTs-650電極的FECH3OH分別為85.8%、78.6%和 72.2%。需要指出的是,實驗中有總FE大于100%的情況出現,這可能是由于實驗誤差導致的。此外由圖4和表1可知,所有產物中CH3OH為主產物,對于TiO2NTs- 450而言,在電解電位為?0.56 V時,FECH3OH是FECO的11.9倍、FEHCOOH的19倍和FEHCHO的40.8倍。
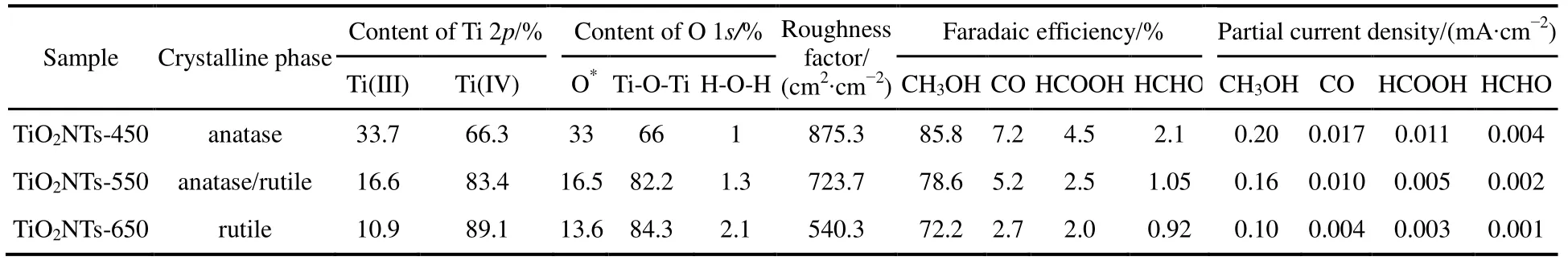
表1 TiO2NTs電極的物理特性及其在電解電位為?0.56 V時的電催化活性Table 1 Summary of physicochemical parameter of various TiO2NTs electrodes at a potential of ?0.56 V.

圖 3 TiO2NTs-450(a)、TiO2NTs-550(b)和TiO2NTs-650(c)電極的FESEM圖Fig.3 FESEM images of TiO2NTs-450 (a),TiO2NTs-550 (b) and TiO2NTs-650 (c) electrodes.
以電極的 FECH3OH/FEH2比值來表征電化學還原 CO2為 CH3OH的選擇性。TiO2NTs-450、TiO2NTs-550和 TiO2NTs-650電極在電解電位為?0.56 V時,其FECH3OH/FEH2分別為34.3、5.7和3.1,則三個電極上產生 CH3OH的選擇性分別為97.2%、85.1%和 75.6%,這說明 TiO2NTs-450電極具有更高的產 CH3OH選擇性。此外,較之TiO2NTs-550和 TiO2NTs-650電極,TiO2NTs-450電極還具有更寬的抑制生成氫的電化學窗口(?0.76 ? ?0.46 V)。
圖 5為 TiO2NTs-450、TiO2NTs-550和TiO2NTs-650電極上總電流密度及各產物分電流密度隨電位變化曲線,本實驗總電流密度隨過電位的增加而增加,當電解電位為?1.16 V時,對應于TiO2NTs-450、TiO2NTs-550和TiO2NTs- 650的總電流密度分別為4.03、2.85和2.13 mA·cm?2。
CO2還原產物的分電流密度隨電極電位變負逐漸增加,當電極電位達到?0.76 V時趨于穩(wěn)定,此電位下PCDCH3OH在TiO2NTs-450、TiO2NTs-550和TiO2NTs-650電極上分別為0.81、0.23和0.16 mA·cm?2,PCDCO分別為 0.039、0.014 和 0.008 mA·cm?2,PCDHCOOH分別為 0.027、0.011和 0.006 mA·cm?2,PCDHCHO分別為 0.006、0.002 和 0.002 mA·cm?2,同時 PCDH2從?0.76 V 時的 0.30、0.24和 0.25 mA·cm?2分別增加至 3.04、2.38和 1.89 mA·cm?2,說明在更負電極電位下析氫副反應增強。
表2為本研究與文獻的PCDCH3OH對比,盡管本文電極生成CH3OH的FE更高,但PCDCH3OH仍然相對較低,這是因為CO2直接還原為CH3OH是復雜的 6電子過程。由于實際工業(yè)應用要求電流密度達到 100 mA·cm?2以上18,所以進一步的工作需在電極幾何面積不變的前提下,極大地提高其電化學活性表面積。
此外,計算產生速率在化工生產中具有重要意義,不同催化劑在不同電勢下CH3OH的產生速率見圖6。由圖可知,三個電極上CH3OH的產生速率均隨過電位的增大而增大,在相同的電極電位下,電極制備過程中煅燒溫度的升高會減慢CH3OH的生成。在研究的電位范圍內,最大CH3OH的產生速率出現在?1.16 V,對于TiO2NTs-450、TiO2NTs-550和 TiO2NTs-650電極而言分別為 5.76、2.70 和 1.49 μmol·cm?2·h?1;而在電極電位為?0.56 V(此時 FE最大)時,CH3OH在TiO2NTs-450、TiO2NTs-550和TiO2NTs-650電極上的產生速率分別為 1.25、1.00和 0.62 μmol·cm?2·h?1。較之文獻報道的 CH3OH 產生速率(表 2),TiO2NTs-450電極優(yōu)于 n-GaAs、p-GaAs和p-InP電極,但劣于CuO電極。鑒于CuO電極存在Cu浸出毒性風險,TiO2NTs較之CuO電極還是有一定優(yōu)越性的。
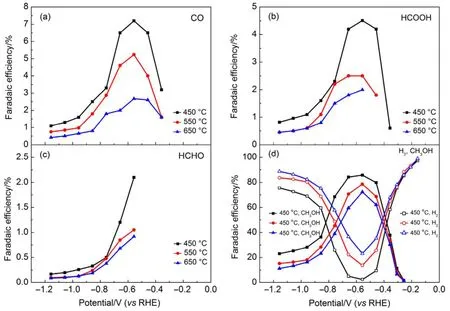
圖4 TiO2NTs-450、TiO2NTs-550和TiO2NTs-650電極上各產物法拉第效率隨電位變化Fig.4 Faradaic efficiency of each CO2 reduction products for TiO2NTs-450, TiO2NTs-550 and TiO2NTs-650 electrodes as a function of cathode potential.(a) CO; (b) HCOOH; (c) HCHO; (d) H2 and CH3OH.
3.3 電化學還原CO2機理
基于TiO2還原CO2的機理主要有兩條途徑,如式(5)和(6)所示19:
CO2→ ·CO2?→ CO → C → ·CH → ·CH3→
CH3OH/CH4(途徑一) (5)CO2→ ·CO2?→ HCOOH → HCHO → CH3OH
(途徑二) (6)
為進一步探明TiO2納米管電極上電化學還原CO2的機理,以CO和HCOOH替代CO2進行120 min電化學反應,CO飽和的0.1 mol·L?1KHCO3溶液用HClO4調節(jié)初始pH為6.8,以確保與CO2飽和的0.1 mol·L?1KHCO3溶液的初始pH一致,而在 0.1 mol·L?1KHCO3溶 液中 加入 75 μL HCOOH控制其初始pH為6.8。實驗結果表明,在通 CO的反應體系中(無 CO2)除 H2外未檢測到其他含碳物質,而單獨添加 HCOOH的反應體系中,產生 CH3OH的法拉第效率和產量分別為75.5%和8.9 μmol,產HCHO的法拉第效率和產量分別為5.9%和1.04 μmol,由此說明本實驗體系中CO是終產物,不會被進一步還原。CH3OH是以HCOOH為中間產物,經途徑二產生的,根據文獻20?22,電化學還原CO2產生CH3OH的具體反應方程如式(7)至(11)。
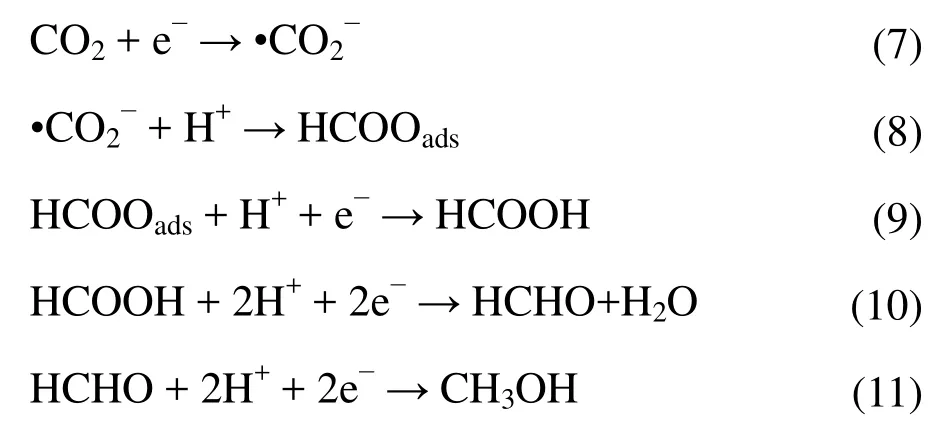
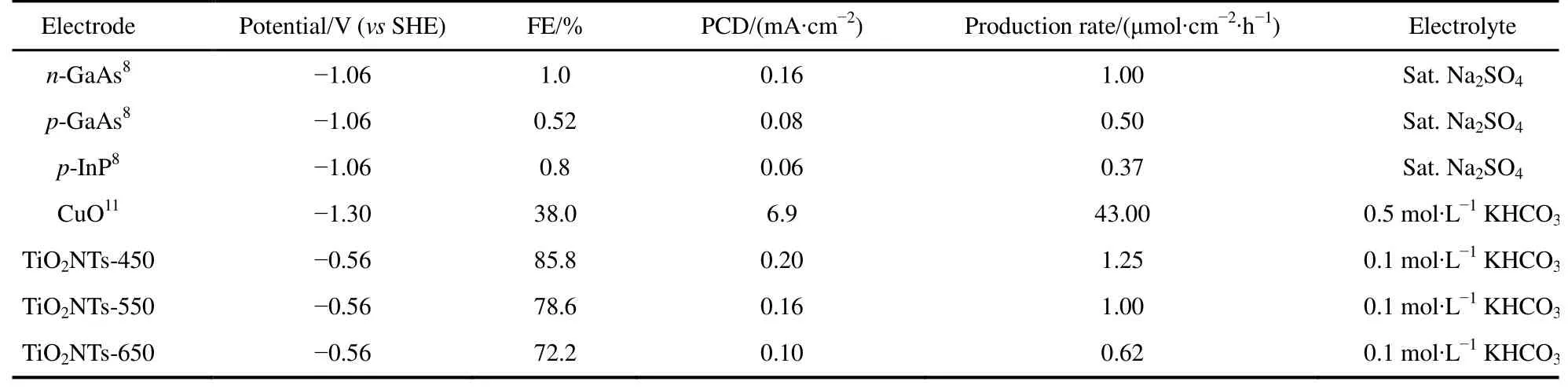
表2 不同半導體電極產甲醇性能比較Table 2 Electrochemical reduction of CO2 to methanol on different semiconductor electrodes.
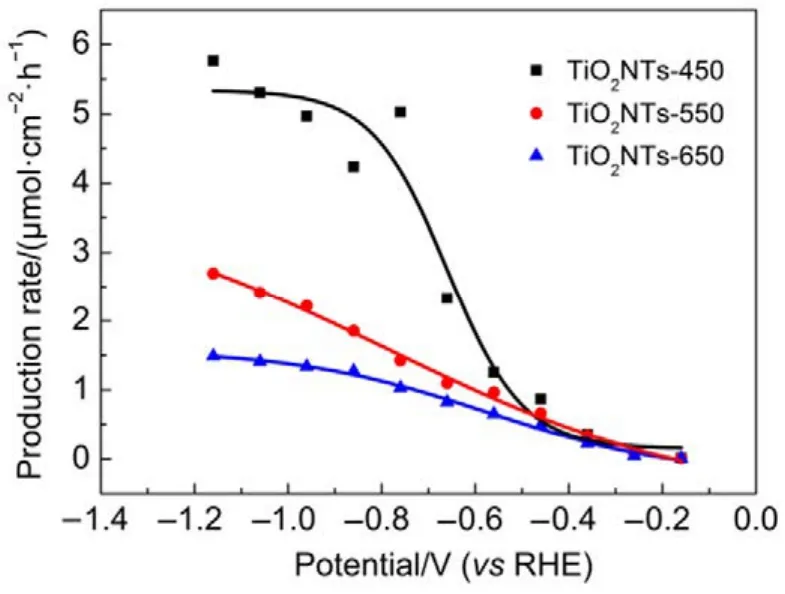
圖6 CH3OH產生速率隨電解位變化Fig.6 Production rate CH3OH with cathode potential.
CO2電化學還原為HCOOH、HCHO和HCHO和CH3OH過程中需要分別傳遞2、4和6個電子,在本實驗中,HCOOH和 HCHO濃度未累積,說明電化學還原HCOOH為HCHO(式(10))和HCHO還原為CH3OH(式(11))不是速率控制步驟。許多研究認為CO2得1電子還原生成·CO2?(式(7))為速率控制步驟23,因其標準電極電位為?1.9 V。然而根據Fletcher24以第一性原理為基礎推導的理論可知,·CO2?得質子反應(式(8))為速率控制步驟。Fletcher推導得到的塔菲爾斜率公式如式(12)。
b = 2.303RT/(afF) (12)
af= np+ nqβf(13)
式中,b為塔菲爾斜率(mV·dec?1),R為理想氣體常數(8.314 J·mol?1·K?1),T 為反應溫度(K),af為傳遞系數,np為速率控制步驟前電子轉移個數,nq為速率控制步驟電子轉移個數(1,或0 (化學控制步驟)),βf為對稱因子(一般取0.5)。
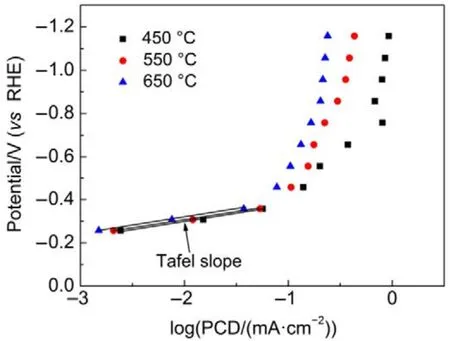
圖7 TiO2NTs-450、TiO2NTs-550和 TiO2NTs-650電極上產CH3OH的塔菲爾曲線Fig.7 Potential versus methanol production partialcurrent density on TiO2NTs-450, TiO2NTs-550 and TiO2NTs-650 electrodes.
基于公式(12)24,當第一電子轉移為控制步驟時,其塔菲爾斜率應為120 mV·dec?1,與本實驗值70 mV·dec?1(圖 7)不一致;而當第二步化學反應為速率控制步驟時,塔菲爾斜率為 60 mV·dec?1與本實驗值接近,這說明·CO2?得質子反應為 TiO2NTs電極上電化學還原CO2的速率控制步驟。這可能是因為Ti(III)參與了CO2的電化學還原過程,從而加速了CO2得電子反應(式(7))。為證明此推斷,考察了三個電極比活性與本征 Ti(III)含量之間的關系,以扣除電化學表面積的影響,具體結果如圖8所示。
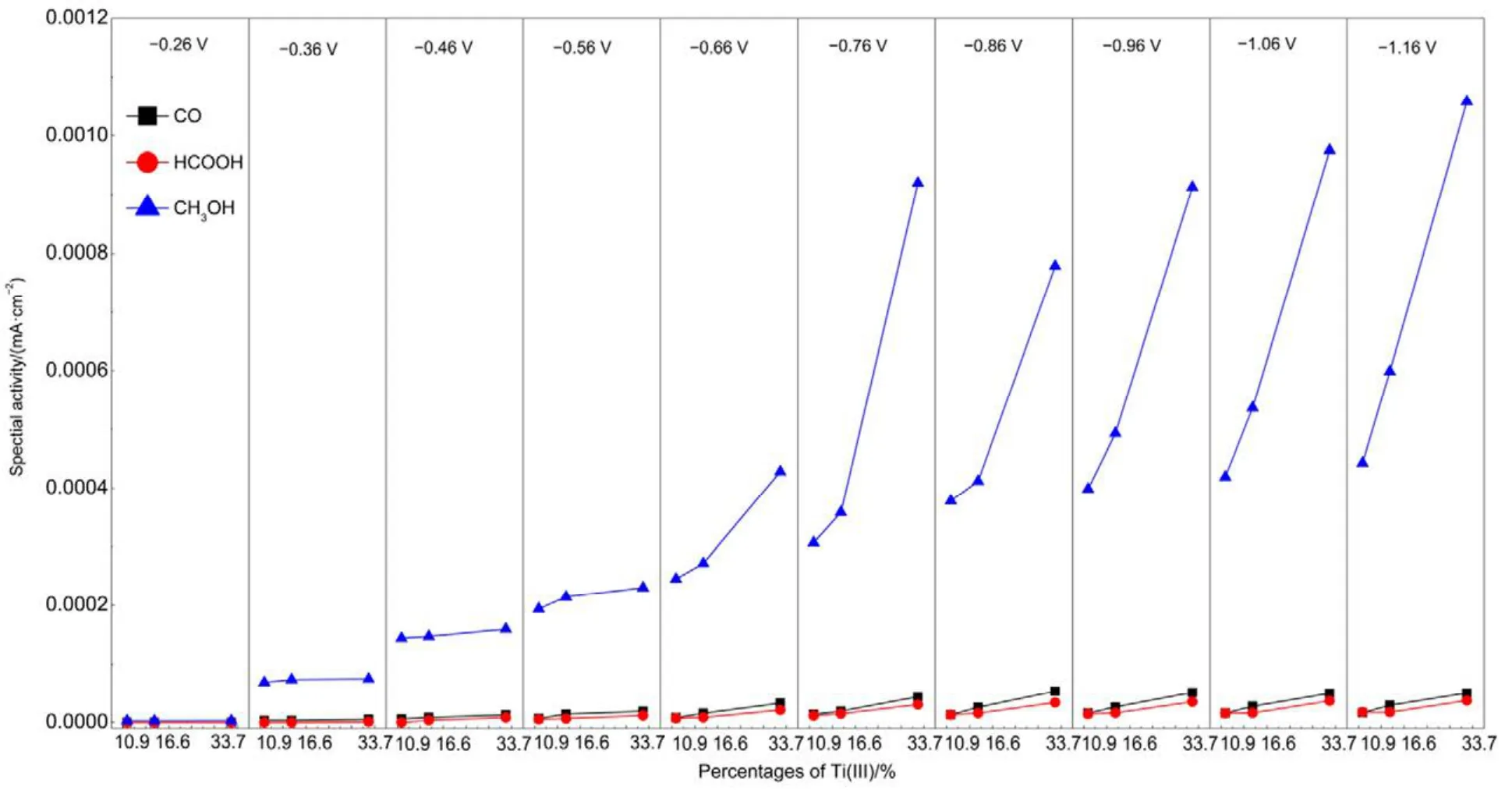
圖8 CO2還原產物的比活性與Ti(III)含量的關系Fig.8 Specific activity of products formed from CO2electroreduction versus percentages of Ti(III).
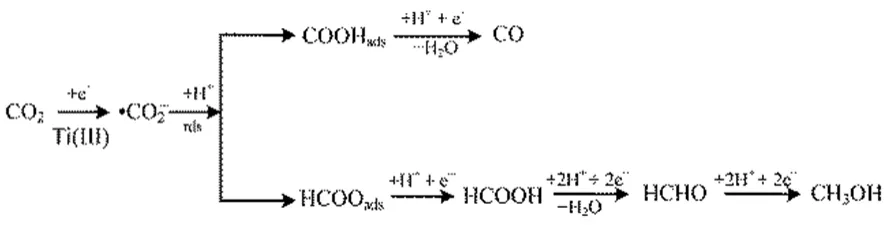
圖9 TiO2NTs電極上CO2電化學還原途徑Fig.9 Proposed reaction pathway for CO2 electroreduction on the TiO2NTs electrode.
由圖8可知,TiO2NTs電極上CO2各還原產物的比活性與 Ti(III)的含量成正相關,說明電極表面的Ti(III)有利于CO2電還原,Weitz等25認為Ti(III)位為弱路易斯酸位,在其鄰位存在相應的強路易斯堿位[O],CO2中的C原子與[O]位結合,從而形成Ti2O-CO2;或者CO2中的C原子直接與Ti3+結合,同時CO2中的O原子占據相鄰的氧空位,從而形成TiO2-COOH。Xiong等26通過CO2-TPD實驗也證明這一結論。因此推斷出TiO2NTs電極上CO2電化學還原為CH3OH的途徑如圖9所示,由于Ti(III)的存在,促進了·CO2?的生成,導致控速步驟轉變?yōu)椤O2?得質子反應。
4 結 論
(1) 通過陽極氧化-高溫煅燒法制備了TiO2NTs電極,采用XRD、XPS、FESEM和雙電位階躍等技術對電極表面結構、形貌及粗糙程度進行表征,結果表明所制備的TiO2NTs電極由管徑約為69?82 nm,管壁厚度為24?27 nm的納米管組成,隨著煅燒溫度的升高,Ti(III)含量降低,且氧空位數量減少。
(2) 電催化實驗結果表明,CO2電化學還原的主要含碳產物為CH3OH、CO、HCOOH和HCHO,其中CH3OH為主產物。在電解電位為?0.56 V時,電化學還原120 min后TiO2NTs-450、TiO2NTs-550和TiO2NTs-650電極上CH3OH的法拉第效率分別為85.8%、78.6%和72.2%,對應的分電流密度為0.2、0.16和0.10 mA·cm?2。
(3) TiO2NTs電極上電化學還原CO2為CH3OH機理研究表明,CH3OH是以HCOOH和HCHO為中間產物生成的。電極表面的Ti(III)含量與電極催化活性成正相關,·CO2?得質子反應為速率控制步驟。
(1) Qiao, J. L.; Liu, Y. Y.; Hong, F.; Zhang, J. J. Chem. Soc. Rev. 2014,43, 631. doi: 10.1039/c3cs60323g
(2) Ritter, S. K. Chem. Eng. News. 2007, 85(18), 11.
(3) Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z. J.; Greeley, J.; Strasser, P.;Cuenya, B. R. J. Am. Chem. Soc. 2014, 136, 16473.doi: 10.1021/ja508879j
(4) He, Z. Q.; Tang, J. T.; Shen, J.; Chen, J. M.; Song, S. Appl. Surf. Sci.2016, 364, 416. doi: 10.1016/j.apsusc.2015.12.163
(5) Liu, Z. F. Acta Phys. -Chim. Sin. 2016, 32, 793. [劉忠范. 物理化學學報, 2016, 32, 793.] doi: 10.3866/PKU.WHXB201602181
(6) Lee, C. H.; Kanan, M. W. ACS Catal. 2014, 5, 465.doi: 10.1021/cs5017672
(7) Olah, G. A.; Goeppert, A.; Prakash, G. K. S. J. Org. Chem. 2008, 74,487. doi: 10.1021/jo801260f
(8) Canfield, D.; Frese, K. W., Jr. J. Electrochem. Soc. 1983, 130, 1772.doi: 10.1149/1.2120090
(9) Summers, D. P.; Leach, S.; Frese, K. W., Jr. J. Electroanal. Chem.1986, 205, 219. doi: 10.1016/0022-0728(86)90233-0
(10) Seshadri, G.; Lin, C.; Bocarsly, A. B. J. Electroanal. Chem. 1994,372, 145. doi: 10.1016/0022-0728(94)03300-5
(11) Le, M.; Ren, M.; Zhang, Z.; Sprunger, P. T.; Kurtz, R. L.; Flake, J.C. J. Electrochem. Soc. 2011, 158, E45. doi: 10.1149/1.3561636
(12) Qu, J. P.; Zhang, X. G.; Wang, Y. G.; Xie, C. X. Electrochim. Acta 2005, 50, 3576. doi: 10.1016/j.electacta.2004.11.061
(13) Zhao, F. M.; Yan, F.; Qian, Y.; Xu, Y. H.; Ma, C. A. J. Electroanal.Chem. 2013, 698, 31. doi: 10.1016/j.jelechem.2013.03.014
(14) Water Quality-Determination of Formaldehyde-Acetylacetone Spectrophotometric Method. GB/T 1397-1991. [水質甲醛的測定—乙酰丙酮分光光度法. GB/T 13197-1991.]
(15) Liu, L. J.; Li, Y. Aerosol. Air Qual. Res. 2014, 14, 453.doi: 10.4209/aaqr.2013.06.0186
(16) Zhang, H.; Chen, J. H.; Chen, H. B.; Lin, C. J. J. Mol. Catal. 2006,20, 249. [張 慧, 陳建華, 陳鴻博, 林昌健. 分子催化, 2006, 20,249.] doi: 10.16084/j.cnki.issn1001-3555.2006.03.013
(17) Wang, W.; Ni, Y. R.; Lu, C. H.; Xu, Z. Z. RSC Adv. 2012, 2, 8286.doi: 10.1039/c2ra21049e
(18) Goeppert, A.; Czaun, M.; Jones, J. P.; Prakash, G. K. S.; Olah, G. A.Chem. Soc. Rev.2014, 43, 7995. doi: 10.1039/c4cs00122b
(19) Chen, X. W. Study on Preparation of Modified TiO2Nanotube Photoelectrode and Its Photoelectrocatalytic Degradation of Diclofenac. Ph. D. Dissertation, Harbin Institute of Technology,Harbin, 2014. [程修文. 改性TiO2納米管光電極制備及光電催化降解雙氯芬酸研究[D]. 哈爾濱: 哈爾濱工業(yè)大學, 2014.]
(20) Russell, P. G.; Kovac, N.; Srinivasan, S.; Steinberg, M. J.Electrochem. Soc. 1977, 124, 1329. doi: 10.1149/1.2133624
(21) Jitaru, M.; Lowy, D. A.; Toma, M.; Toma, B. C.; Oniciu, L. J. Appl.Electrochem. 1997, 27, 875. doi: 10.1023/a:1018441316386
(22) Innocent, B.; Pasquier, D.; Ropital, F.; Hahn, F.; Léger, J. M.;Kokoh, K. B. Appl. Catal. B2010, 94, 219. doi:10.1016/j.apcatb.2009.10.027
(23) Hori, Y.; Murata, A.; Takahashi, R. J. Chem. Soc., Faraday Trans.1989, 85, 2309. doi: 10.1039/F19898502309
(24) Fletcher, S. J. Solid State Electrochem. 2009, 13, 537.doi: 10.1007/s10008-008-0670-8
(25) Bhattacharyya, K.; Danon, A. K.; Vijayan, B. K.; Gray, K. A.; Stair,P. C.; Weitz, E. J. Phys. Chem. C 2013, 117, 12661.doi: 10.1021/jp402979m
(26) Xiong, L. B.; Li, J. L.; Yang, B.; Yu, Y. J. Nanomater. 2012, 2012,1. doi: 10.1155/2012/831524
Electrochemical Reduction of CO2to Methanol at TiO2Nanotube Electrodes
QIU Jian-Ping1,2TONG Yi-Wen1ZHAO De-Ming3HE Zhi-Qiao1CHEN Jian-Meng1SONG Shuang1,*
(1College of Environment, Zhejiang University of Technology, Hangzhou 310032, P. R. China;2Jinhua Polytechnic, Jinhua 321007, Zhejiang Province, P. R. China;3College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310032, P. R. China)
A series of highly ordered TiO2nanotube (TiO2NTs) electrodes are prepared via potentiostatic anodization of Ti foil followed by calcining in air. X-ray diffraction (XRD), field-emission scanning electron microscopy (FESEM), X-ray photoelectron spectroscopy (XPS), and potential steps determination are used to characterize the electrodes. The electrochemical reduction of CO2on these three TiO2NTs electrodes is investigated by cyclic voltammetry and potentiostatic electrolysis in 0.1 mol·L?1KHCO3aqueous solution. Methanol is found to be the major product in electrochemical CO2reduction, while formic acid, formaldehyde, methane, and CO are formed as minor products. Compared with the electrodes sintered at 550 and 650 °C, the optimal TiO2NTs electrode is found to be the one calcined at 450 °C (TiO2NTs-450). After 120 min of reaction, the Faradaic efficiency and partial current density of methanol is 85.8% and 0.2 mA·cm?2at ?0.56 V vs reversible hydrogen electrode (RHE), respectively. The trivalent titanium in TiO2serves as an efficient site for adsorption of CO2and stabilization of the adsorbed ·CO2?radical. Consequently, the reduction of CO2on TiO2NTs electrodes involves a fast firstelectron and proton transfer followed by a slow second proton transfer as the rate-limiting step.
TiO2nanotube electrode; Electrochemical reduction; CO2; Methanol;Reaction mechanism
December 21, 2016; Revised: March 24, 2017; Published online: April 7, 2017.
O646
10.3866/PKU.WHXB201704078 www.whxb.pku.edu.cn
*Corresponding author. Email: ss@zjut.edu.cn; Tel: +86-571-88320726.
The project was supported by the National Natural Science Foundation of China (21477117) and Natural Science Foundation of Zhejiang Province, China(LR14E080001, LQ15E080007, LY15B070005).
國家自然科學基金(21477117)和浙江省自然科學基金(LR14E080001, LQ15E080007, LY15B070005)資助項目
? Editorial office of Acta Physico-Chimica Sinica
