乙酰膽堿酯酶AChE與1,7-二氮雜咔唑抑制劑的作用機理的分子動力學模擬
2017-11-13 12:22:26趙騰騰楊雪雨董珂珂朱小蕾
無機化學學報 2017年11期
趙騰騰 楊雪雨 董珂珂 朱小蕾
乙酰膽堿酯酶AChE與1,7-二氮雜咔唑抑制劑的作用機理的分子動力學模擬
趙騰騰 楊雪雨 董珂珂 朱小蕾*
(南京工業大學化工學院,材料化學工程國家重點實驗室,南京 210009)
通過分子對接、分子動力學(MD)模擬以及成鍵自由能分析方法,從原子水平上模擬研究了3種1,7-二氮雜咔唑衍生物(分別記為M1、M2和M3)與AChE的結合模式及相互作用機理,分析和討論了研究體系的靜電相互作用和范德華相互作用(vdW)。用MM-PBSA方法計算的3種抑制劑與AChE之間的結合自由能與抑制劑的實驗生物活性數據(IC50值)相對應。分析結果表明,殘基S286與抑制劑之間形成的氫鍵作用有利于抑制劑與AChE之間的結合。范德華相互作用,尤其是抑制劑與關鍵殘基W279和Y334的作用,對抑制劑與AChE之間的結合自由能有較大的貢獻,在區分抑制劑M1(或M2)和M3的生物活性上發揮著重要的作用。
阿爾茨海默癥;乙酰膽堿酯酶;1,7-二氮雜咔唑類抑制劑;分子動力學模擬;MM-PBSA
眾所周知,世界正面臨老齡化問題,阿爾茨海默病(AD)的發病率隨著年齡的增加而顯著增加。阿爾茨海默癥病人的主要表現為認知功能障礙[1]、記憶缺失及語言障礙[2-3]等神經精神癥狀。迄今為止,阿爾茨海默病本質上可能的病因是基底前腦膽堿能神經遞質的缺陷,尤其是乙酰膽堿(ACh)的大量缺失[4-6]。膽堿酯酶包括乙酰膽堿酯酶(AChE)和丁酰膽堿酯酶(BChE),其主要作用是使膽堿能神經遞質水解。在ACh的水解酶作用下[7],ACh將被水解為膽堿和乙酸,從而引起阿爾茨海默癥。因此,抑制AChE和BChE被認為是最有前途的治療阿爾茨海默病的策略。
根據X射線結構分析[8],AChE的活性位點中存在一個狹而深的峽谷,包括兩個不同的結合位點:位于峽谷底部的Ser-His-Glu催化位點(CAS),以及位于峽谷入口的外圍陰離子結合位點(PAS)[8]。 因此,AChE抑制劑可分為兩種類型,即單位點抑制劑和雙位點抑制劑[8-12]。前人研究[10,13]表明,與單位點抑制劑相比,雙位點抑制劑能更有效的治療AD。
至今,實驗上已經設計合成了許多AChE和BChE抑制劑。例如,Xie等[14]設計合成了一系列新型的具有多功能多靶點的香豆素類混合抑制劑,其中大多數抑制劑能有效地抑制AChE的活性。Alpan等[15]設計合成了苯并咪唑及其衍生物類抑制劑,并進行了活性分析,發現改變雜環上的親脂性原子或者環上的取代基均可增強AChE抑制劑的活性。Maryam等[16]合成了一系列的新型的吖啶酮連接到1,2,3,-三唑的衍生物,并預測了它們對AChE和BChE的生物活性,對接結果與實驗結果相一致。Asadipour等[17]設計合成了一系列由香豆素、甲酰胺基以及芐基哌啶這幾個結構組成的芐基哌啶類AChE抑制劑,對接研究表明它們具有雙重結合位點。Gao 等[18]和 Liu 等[19]設計合成了一系列新型的氯查耳酮的叔胺衍生物,并評估了這一類衍生物對AChE和BChE的生物活性。最近,Gazzard等[20]發現,1,7-二氮雜咔唑衍生物是AChE強有效的抑制劑。在理論研究方面,Niu等[21]采用拉伸分子動力學模擬方法研究了抑制劑E2020與AChE的結合機制,并在原子水平上為E2020從乙酰膽堿酯酶 (TcAChE)的解離提供了新的思路。Puiatti等[22]通過分子對接及分子動力學模擬對抑制劑與AChE的結合模式進行了研究,并比較了3種石松類生物堿抑制劑與AChE的結合模式,通過MM-GBSA方法計算的3個復合物的結合能與實驗上抑制劑的IC50值相一致。Zhu等[23]利用分子動力學和MM-PBSA方法,研究了syn1-TZ2PA6和anti1-TZ2PA6兩種同分異構體分別對AChE1和AChE2的選擇性機制。Zhou等[24]通過3DQSAR方法、分子對接和分子動力學模擬對60種他克林衍生物及其對AChE抑制活性的影響進行了研究。結果表明,AChE與抑制劑形成的復合物的空間位阻和氫鍵對抑制劑的生物活性影響較大。AChE的結合位點的關鍵殘基包含 Tyr70、Trp84、Tyr121、Trp279 和Phe330。Shi等[25]用分子對接和分子動力學模擬方法,研究了對映體(RS,S)-17b 和(RS,R)-17b 與 AChE 的結合模式,為設計新型高效的AChE抑制劑提供了新的線索。最近,Mohammadi等[26]通過改變烷基取代基設計出了許多氨基甲酸酯衍生物,并利用分子對接和分子動力學模擬方法研究了取代結構對抑制劑與AChE結合能以及AChE構象變化的影響。Hossain等[27]在化學計量學和動力學模擬分析的基礎上研究了乙酰膽堿酯酶抑制劑對分子結構的要求。
目前許多AChE抑制劑已經在臨床上應用于AD的治療,例如他克林(Tacrine)[28]、多奈哌齊(Donepezil)[29-30]、加蘭他敏(Galanthamine)[31]、利凡斯的明(Rivastigmine)[32-33]等藥物,但是治療中仍然伴隨著一定的副作用。上述問題出現的主要原因之一是抑制劑與AChE之間的作用機理還沒有清楚地理解,這是一個亟待解決的問題。因此,設計高效的最小副作用的新型AChE抑制劑仍然是非常重要且具有挑戰性的任務。目前,從原子層次上揭示抑制劑與AChE詳細的作用機理還沒有報道,而這些研究的進行有利于解決上述問題以及加速實驗研究。在這個過程中,分子動力學模擬是一個關鍵性的技術[21-27]。因此,我們選擇了3個1,7-二氮雜咔唑衍生物作為AChE抑制劑,利用分子對接、分子動力學模擬、自由能計算以及能量分解等方法來研究抑制劑與AChE的結合機理,揭示AChE抑制劑的生物活性大小的原因。模擬結果證明了范德華相互作用(vdW)對結合自由能有較大的貢獻,靜電相互作用(包括氫鍵作用)有利于3個抑制劑穩定在AChE的結合位點中。結合位點上的主要關鍵殘基 (W279和Y334),在抑制劑與AChE的結合中以及區分3個抑制劑的生物活性上發揮著重要作用。本文的研究為今后設計更高效的AChE抑制劑提供了理論指導。
1 實驗部分
1.1 準備初始結構
AChE的X射線晶體結構是從PDB數據庫(PDB代碼:1EVE,分辨率為 0.25 nm)[34]中提取的,去除該晶體結構中的配體、結晶水分子和氫原子,作為初始的蛋白質結構。利用畫圖軟件構建3個1,7-二氮雜咔唑衍生物(分別標記為M1、M2和M3),如表1中所示,并在B3LYP/6-31G*水平上用Gaussian 09軟件[35]進行優化得到最優結構,優化后的結構作為分子對接所需配體的初始結構。
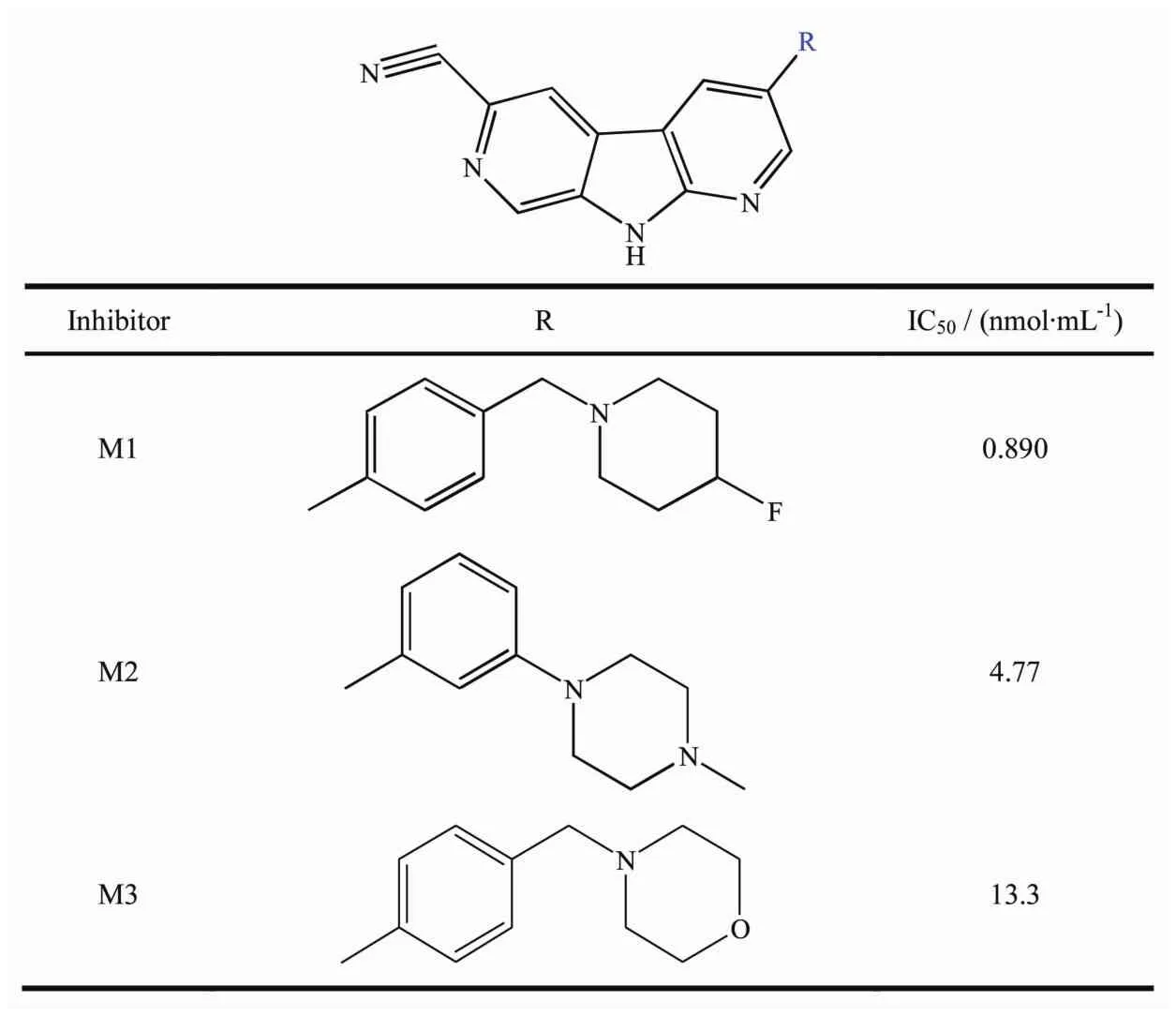
表1 3個1,7-二氮雜咔唑衍生物抑制劑的結構和IC50值Table 1 Structures and bioactivity of three 1,7-diazacarbazole derivative inhibitors to AChE
1.2 對接方法
小分子配體與受體的相互作用首先通過分子對接研究。我們采用Autodock4.0軟件的拉馬克遺傳算法[36]進行了分子對接的研究,將極性氫原子添加到蛋白質的各個殘基上,再將科爾曼聯合原子電荷添加到AChE的部分原子上。在對接過程中,將受體的格點盒子大小設置為6.5 nm×6.5 nm×6.5 nm,間隔為0.037 5 nm。用遺傳算法共進行200次獨立對接計算,然后從對接構象的最大簇(均方根偏差RMSD設定為0.2 nm)選出最優構象,最終得到的復合物體系的結構用作后續的MD模擬的初始結構。
1.3 分子動力學模擬
我們采用AMBER10.0軟件分別對3個復合物體系的進行MD模擬,本文中受體和配體的力場分別為AMBERFF03[37]和 GAFF[38]力場,用 Antechamber模塊添加配體的氫原子并擬合RESP(限制靜電勢)[39]電荷。將3個復合物體系分別加到一個溶劑為TIP3P水分子模型[40]的盒子中,盒子半徑為1.0 nm。氫原子通過LEaP模塊添加,并添加抗衡離子Na+使體系電荷呈中性。在MD之前,對每個體系進行能量最小化,采用 2 000步的最小化法和2 000步的共軛梯度法,非鍵截斷半徑設為1.0 nm。MD模擬過程先進行200 ps的41 840 kJ·mol-1·nm-2限制性加熱(0~300 K),然后在 NPT 系綜(P=101 kPa,T=300 K)下進行時間步長為0.002 ps的50 ns非限制的MD模擬。在MD模擬過程中,采用SHAKE算法[41]對原子鍵長進行約束,長程靜電相互作用采用 Particle-mesh Ewald (PME)[42]進行處理。
1.4 結合自由能計算
為了清楚地了解3個抑制劑與AChE的結合機制,我們通過AMBER10.0中的MM-PBSA程序[43]計算得到每個復合物體系的結合自由能以及能量分解。對于每個體系,取穩定后的最后5 ns的MD軌跡,提取200個構象對抑制劑與AChE的結合自由能計算來研究生物活性。配體與受體結合自由能(ΔGbind)可表述為[44-45]:
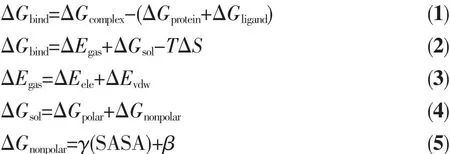
其中ΔGcomplex、ΔGprotein和ΔGligand分別代表復合物、蛋白質以及配體的自由能。結合自由能又可分解為三部分:氣相結合能(ΔEgas)、溶劑化自由能(ΔGsol)和熵貢獻(-TΔS)(式 2)。 ΔEgas又可進一步分解成靜電能(ΔEele)和范德華作用能(ΔEvdW)(式 3)。 溶劑化自由能(ΔGsol)可以分為兩部分:極性溶劑化自由能(ΔGpolar)和非極性溶劑化自由能 (ΔGnonpolar)(式 4)。 非極性溶劑化自由能(ΔGnonpolar)通過計算方程 (式5)中的溶劑可及表面積(SASA)獲得,其中SASA可以通過AMBER 10.0軟件中的Molsurf方法得到。溶劑的探針半徑設為0.14 nm。對應的溶劑參數γ和β分別設為0.226 77 kJ·mol-1·nm-1和 3.85 kJ·mol-1。 溶質和溶劑的介電常數分別設為1.0和80.0。另外,由于熵的計算對于大體系而言異常耗時,并且對于相似的蛋白質來說小分子的結合引起的熵變很接近[46]。因此,在本文的工作中忽略了熵(-TΔS)對 ΔGbind的貢獻[46-47]。用這種方法對能量的計算在前人的研究工作中也被證實是合理的[48]。
2 結果與討論
2.1 結合模式
在本文中,我們分別將抑制劑M1、M2和M3對接到AChE中,得到最優的對接構象如圖1(a)所示,將用于后續的MD模擬。從圖1(a)中可以看出,3個抑制劑與AChE的結合模式大致相同,詳細的對接模式如圖2所示。在圖2中可以看出,所有抑制劑都位于由殘基 Y70(Tyr70)、V71(Val7l)、D72(Asp72)、S81(Ser81)、W84(Trp84)、N85(Asn85)、G118(Gly118)、Y121(Tyr121)、S122 (Ser122)、W279 (Trp279)、L282 (Leu282)、S286(Ser286)、I287 (Ile287)、F288 (Phe288)、R289(Arg289)、F290 (Phe290)、F330 (Phe330)、F331 (Phe331)、Y334(Tyr334)和 W432(Trp432)形成的結合口袋中。 同時,我們還可以看出3個抑制劑均與AChE形成了氫鍵,使抑制劑與AChE結合模式更加牢固。選擇結合能較低且構象較多的結構作為最好的對接構象,用作后續MD模擬的初始結構。如圖1(b)和圖3所示,在MD模擬之后,3個復合物體系的構象與圖1(a)和圖2中的對接結果很相似,配體周圍的殘基變化不大。每個體系的回旋半徑基本上都能保持穩定。因此,抑制劑與AChE結合時,AChE的結構都是穩定的。
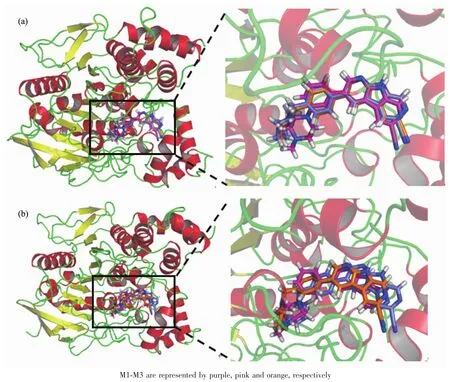
圖1 抑制劑與AChE的結合模式:(a)3個AChE/M1~M3體系的對接構象;(b)3個AChE/M1~M3體系的MD構象Fig.1 Binding modes of the inhibitors with AChE:(a)Docking conformations of three AChE/inhibitor complexes;(b)MD conformations of three AChE/inhibitor complexes
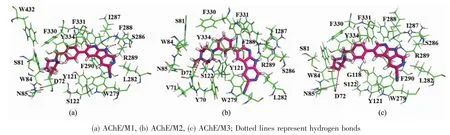
圖2 AChE與3個1,7-二氮雜咔唑類抑制劑復合物Fig.2 Detailed docking binding modes of the three complexes
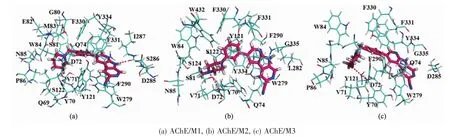
圖3 3個復合物體系MD模擬后的最低能量構象Fig.3 Conformations with lowest energy after MD simulations for three complexes
為了深入了解結合態AChE殘基的變化,我們對蛋白質的柔性進行了分析,蛋白質的柔性可由每一個殘基的B因子(B-factor)來評判。本文我們計算了3個復合物體系的蛋白質殘基的B-factor值,并與游離態AChE的B-factor進行比較。通過分析,3個復合物體系具有相似的B-factor值分布,并且結合態AChE的殘基與游離態AChE實驗晶體結構的B-factor值分布基本一致,這也說明3個抑制劑與AChE的結合模式比較相似。
2.3 AChE與抑制劑的結合機理及抑制劑的生物活性
抑制劑與AChE的結合自由能計算是衡量抑制劑活性的重要方法。本文我們用MM-PBSA的方法計算了3個復合物體系的結合自由能以及不同能量項,結果列在表2中。表2中的能量值,正數表示這種作用不利于抑制劑與AChE的結合,負數表示這種作用使體系能量降低,有利于結合,負數絕對值越大,表示這種作用對結合的貢獻越大。從表2可以看出,M1、M2、M3與 AChE的結合自由能分別為:-144.35、-129.37、-102.47 kJ·mol-1,與實驗中抑制劑生物活性(IC50值分別為 0.890、4.77、13.3 nmol·ml-1)[20]的大小次序相一致,表明抑制劑的生物活性順序:M1>M2>M3。
為了進一步了解每個殘基在結合過程中的作用,我們用MM-PBSA方法對3個復合物體系中(AChE/M1~AChE/M3)每個殘基的結合自由能進行了能量分解,如圖 5(a)所示,標注出的是 ΔGbind小于約-3.5 kJ·mol-1的殘基,可以看出AChE與抑制劑相互作用的關鍵殘基有:AChE/M1(Y70、V71、W84、W279、S286、I287、Y334)、AChE/M2(V71、W84、W279、F331、Y334)、AChE/M3 (V71、Q74、W84、N85、W279、F290、F330、F331、Y334),這與實驗上[20]以及其它理論研究[24]的結果相一致。表2中比較了每一個能量項 (ΔEele、ΔEvdW、ΔGpol、ΔGnonpolar)對總的自由能(ΔGbind)的貢獻。通過分析表2可以看出,范德華作用(ΔEvdW)對AChE與抑制劑的結合起主導作用(-241.17、-234.72、-200.58 kJ·mol-1)。 極性溶劑化能(ΔGpol)對結合是不利的,盡管氣相的靜電能(ΔEele)對結合是有利的,但也不能完全抵消極性溶劑化自由能對結合的不利影響[50-51]。 另外,非極性溶劑化能(ΔGnonpolar)也有較小的貢獻。
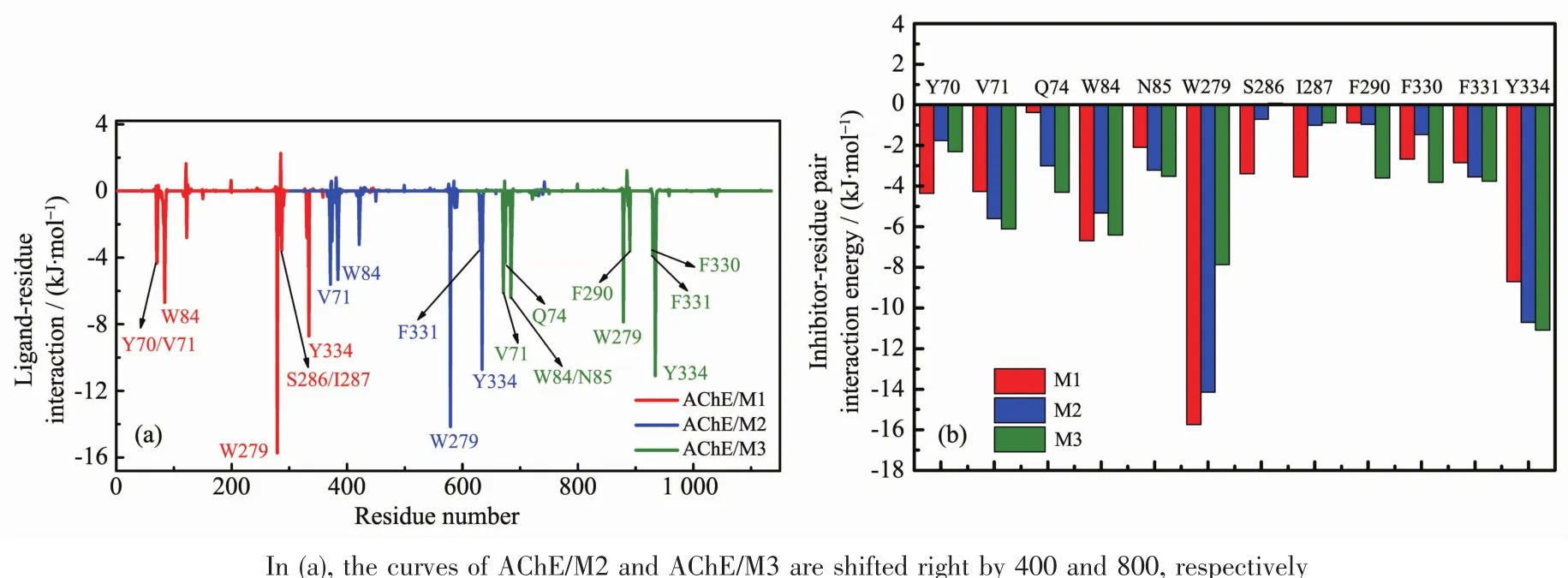
圖5 AChE/抑制劑復合物體系中抑制劑-殘基之間的相互作用能譜 (a)和3個復合物體系的主要殘基(Y70、V71、Q74、W84、N85、W279、S286、I287、F290、F330、F331、Y334)的能量比較 (b)Fig.5 Decomposition of total interaction energy on a per-residue basis for the three proteininhibitor complexes (a)and comparison of interaction energies of the major residues(Y70,V71,Q74,W84,N85,W279,S286,I287,F290,F330,F331,and Y334)for the three AChE/inhibitor complexes(b)
為了更好地驗證3個抑制劑的生物活性,如圖5所示,我們深入分析了關鍵殘基與抑制劑之間的相互作用。從圖 5(a)中可以看出,對于 AChE/M1、AChE/M2、AChE/M3復合物體系,這些關鍵殘基包括Y70、V71、Q74、W84、N85、W279、S286、I287、F290、F330、F331 和Y334。 在這些殘基中,主要殘基 W84、W279、Y334與抑制劑(M1、M2、M3)結合自由能的總和分別為-31.13、-30.17、-25.36 kJ·mol-1,使3個抑制劑的生物活性順序為 M1>M2>M3。 圖 5(b)表示關鍵殘基與 3 個抑制劑的結合自由能圖,殘基Y334與抑制劑的結合自由能明顯弱于殘基W279與抑制劑間的結合自由能,后者更能有效地區分3個抑制劑的生物活性。
通過表2得知,范德華作用對復合物的結合起主導作用,我們對范德華作用進行了殘基自由能分解,結果如圖 6(a)所示,列出的是 ΔEvdW小于約-3.5 kJ·mol-1的殘基。由表2可以看出,抑制劑M1與AChE之間的范德華相互作用(ΔEvdW:-241.17 kJ·mol-1)明顯強于抑制劑M2(或M3)與AChE之間的范德華相互作用(-234.72 (或-200.58)kJ·mol-1),使得 3 個抑制劑的生物活性順序為 M1>M2>M3。如圖 6(a)所示,抑制劑 M1與 關 鍵 殘 基 (Y70、V71、D72、S81、W84、Y121、W279、F330、F331、Y334)之間的范德華相互作用,尤其是殘基Y70、W84、W279、Y334,對結合自由能有較大的貢獻。從圖6(a)可以看出,盡管抑制劑M2和M3與殘基Y334之間的范德華相互作用相對更強,但是抑制劑M1與殘基W279之間的范德華相互作用遠遠強于M2(或M3)與W279之間的范德華相互作用。抑制劑M1、M2、M3 與 這 些 關 鍵 殘 基 (Y70、V71、D72、Q74、S81、W84、Y121、W279、F330、F331、Y334)范德華相互作用之和分別為-84.10、-85.22、-70.12 kJ·mol-1, 與 3個抑制劑的生物活性大小順序(M1(或 M2)>M3)相一致。以上分析表明了抑制劑與殘基之間的范德華相互作用,尤其是殘基W279和Y334,對結合自由能以及區分3個抑制劑的生物活性上有很大的貢獻。
為了揭示靜電相互作用對抑制劑M1、M2、M3與AChE結合親和力的影響,我們用LIGPLOT[52]程序分析了抑制劑與AChE之間的氫鍵作用和疏水相互作用。如圖7所示,結合態AChE周圍的殘基與結合自由能殘基分解分析中得到的殘基是一致的。從圖7(a)中可以看出,在AChE/M1體系中,存在一條氫鍵,形成于殘基 S286的氧原子(O)與 M1上的氮原子(N9)之間。如表3所示,這條氫鍵的占有率高達90.36%,從圖7(d)可以看出,這條氫鍵(O(S286)-N9(M1)在 MD 模擬過程中是趨于穩定的,平均距離是0.295 nm(表3)。在AChE/M2體系中,如圖 7(b)所示,在殘基 D72上的氮原子(N)與M2上的氮原子(N25)之間形成了一條占有率為31.56%的氫鍵,這條穩定的氫鍵 (N(D72)-N25(M2)(圖 7(d)的平均距離為 0.334 nm(見表 3)。同樣的,在AChE/M3體系中,如圖 7(c)所示,殘基 D72上的氮原子(N)與 M3 上的氧原子(O26)之間形成了氫鍵,占有率為 40.94%,這條氫鍵(N(D72)-O26(M3)穩定后的平均距離是0.323 nm(見表3)。為了考察每個殘基的靜電相互作用對抑制劑與AChE結合的細節,我們對靜電相互作用進行了殘基自由能分解,結果如圖6(b)所示,列出的是 ΔEele小于約-3.5 kJ·mol-1的殘基(Y70、D72、E73、E199、D276、D285、S286)。 從圖 6(b)可以看出,雖然M3與殘基D72之間的靜電相互作用很強,但仍比M1與殘基S286之間的靜電相互作用要弱,這與上述的氫鍵分析結果是一致的。抑制劑M1、M2、M3分別與關鍵殘基(D72 和 S286)的凈靜電相互作用(ΔEele+ΔGpol)之和分別為3.60、9.92和10.17 kJ·mol-1,雖然不利于抑制劑與AChE的結合,但與抑制劑的生物活性相一致。上述分析表明,M1與殘基S286之間形成的氫鍵有利于抑制劑穩定在結合位點。

圖6 3個抑制劑與AChE復合物抑制劑-殘基的范德華作用 (a)和3個抑制劑與AChE復合物抑制劑-殘基的靜電相互作用 (b)Fig.6 Van der Waals interaction energy spectra of inhibitor-residue pair in AChE/inhibitor complexes (a)and electrostatic interaction energy spectra of inhibitor-residue pair in AChE/inhibitor complexes (b)
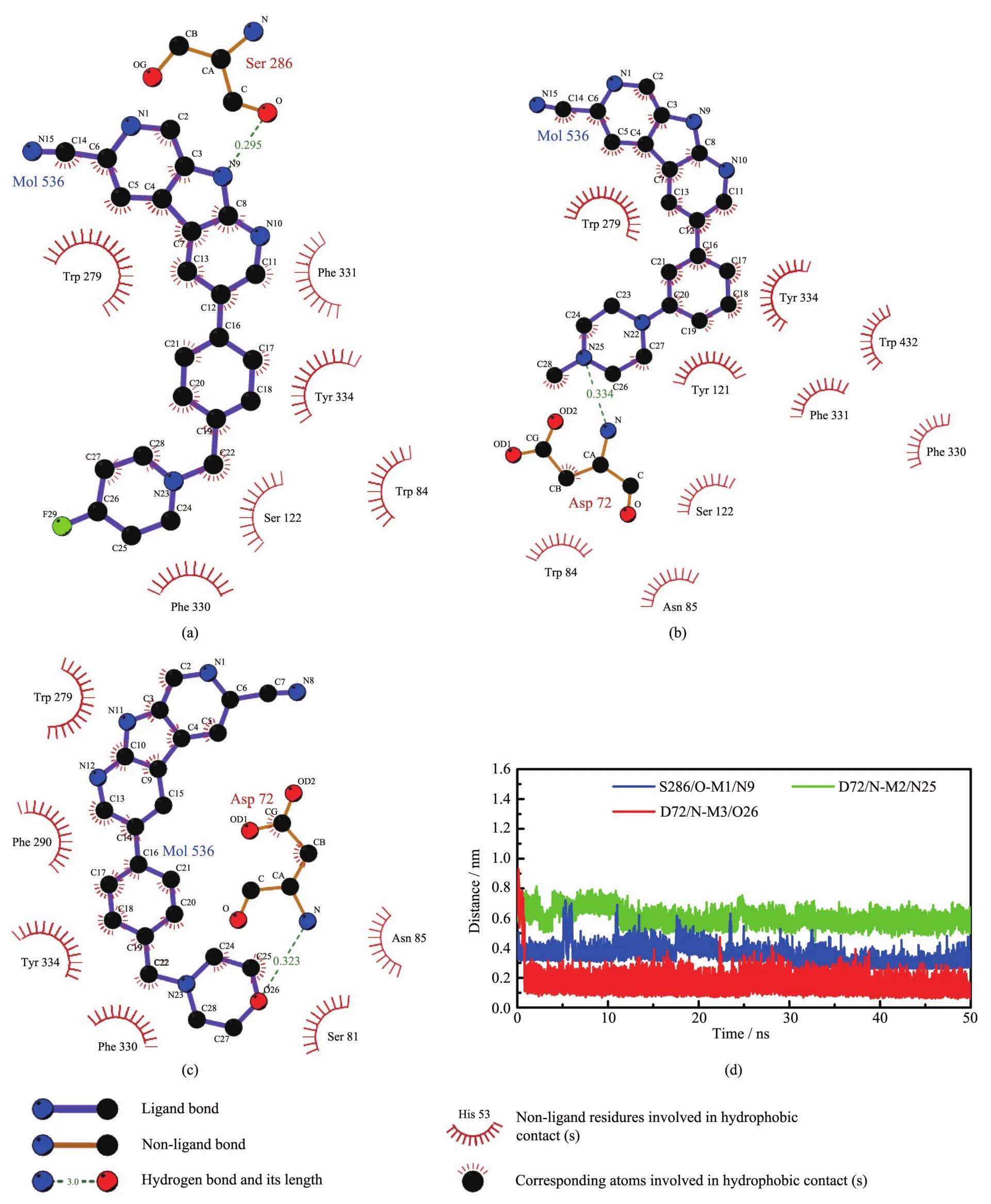
圖7 氫鍵和疏水作用二維圖:虛線代表氫鍵,穗狀代表與3個抑制劑形成疏水作用的殘基(a)AChE/M1;(b)AChE/M2;(c)AChE/M3;(d)抑制劑與AChE之間形成的氫鍵的距離隨MD模擬的時間變化,其中,M1與殘基S286的距離曲線上移了0.05 nm;M2與殘基D72的距離曲線上移了0.2 nmFig.7 2D representation of hydrogen bonds and hydrophobic interactions:Dashed lines represent hydrogen bonds,and spiked residues form hydrophobic interactions for complexes AChE/M1 (a),AChE/M2 (b),and AChE/M3 (c);(d)Interatomic distances with the MD time evolution show the stability of hydrogen bonds between three inhibitors and AChE;M1:Curve of S286 is shifted upward by 0.05 nm;M2:Curve of D72 is shifted upward by 0.2 nm
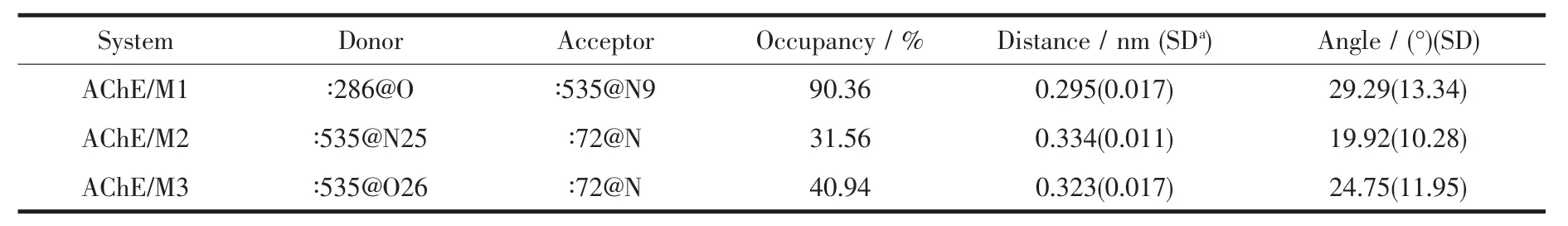
表3 MD模擬過程中抑制劑與AChE之間形成的氫鍵Table 3 Hydrogen bonds formed between inhibitors and AChE during MD simulations
基于上述分析,我們從表 2、圖 6(a)、圖 7 中可以看出,在AChE/M1~M3體系中,抑制劑與AChE之間有較強的范德華相互作用,特別是AChE上的殘基W279與抑制劑二氮雜咔唑母體、殘基Y334與抑制劑上苯環的關鍵作用。抑制劑M1、M2、M3與這2個關鍵殘基 (W279和Y334)的范德華相互作用之和分別為-31.17、-34.60、-24.10 kJ·mol-1。另外,抑制劑 M1 與殘基S286之間的靜電相互作用要強于抑制劑M2/M3與殘基D72之間的靜電相互作用。通過比較AChE/M1、AChE/M2、AChE/M3復合物體系的結構和相互作用,可以看出母體二氮雜咔唑的重要性。如果二氮雜咔唑與含有一個苯環的適當的取代基相連,像抑制劑M1一樣,就會在殘基W279與抑制劑二氮雜咔唑母體之間產生較強的范德華相互作用,在殘基S286與二氮雜咔唑母體上的唑氮之間產生較強的氫鍵作用,形成的二氮雜咔唑衍生物抑制劑對AChE就可能有較強的抑制活性。上述分析揭示了殘基S286對M1與AChE的結合起有利作用,主要關鍵殘基W279和Y334在抑制劑與AChE的結合中和區分3個抑制劑的生物活性中發揮了重要的作用。
3 結 論
本文通過分子對接、分子動力學模擬和MMPBSA能量計算方法,從原子水平上研究了3個1,7-二氮雜咔唑衍生物抑制劑與AChE的相互作用,并揭示了1,7-二氮雜咔唑衍生物抑制劑對AChE的抑制機理和3個抑制劑之間的生物活性差別。模擬結果表明復合物中穩定的氫鍵可以使抑制劑與AChE之間產生較強的靜電作用,有利于兩者的結合。結合能的計算結果與抑制劑的實驗生物活性數據(IC50)吻合較好,證實了抑制劑的生物活性順序為M1>M2>M3。結果表明,抑制劑與殘基S286之間形成的氫鍵作用有助于抑制劑與AChE的結合。范德華相互作用對AChE/M1~M3體系的結合自由能有很大的貢獻。主要關鍵殘基W279和Y334對于抑制劑與AChE的結合及區分3個抑制劑的生物活性大小起了決定性作用。本文的工作有助于理解AChE抑制劑的抑制機理,并為設計新型的有潛力的AChE抑制劑提供了有價值的信息。
[1]Mussele S V D,Bastard N L,Vermeiren Y,et al.Int.J.Geriatr.Psychiatr.,2013,28(3):265-275
[2]Sperling R A,Dickerson B C,Pihlajamaki M,et al.Neuromol.Med.,2010,12(1):27-43
[3]Nestor P J,Fryer T D,Hodges J R.NeuroImage.,2006,30(3):1010-1020
[4]Bartus R T,Dean R L,Beer B,et al.Science,1982,217(4558):408-414
[5]Terry R D,Masliah E,Salmon D P,et al.Ann.Neurol.,1991,30(4):572-580
[6]Francis P T,Palmer A M,Snape M,et al.J.Neurol.Neurosur.Ps.,1999,67(4):558-558
[7]Talesa V N.Mech.Ageing Dev.,2001,122(16):1961-1969
[8]Alipour M,Khoobi M,Moradi A,et al.Eur.J.Med.Chem.,2014,82:536-544
[9]Liu S,Shang R,Shi L,et al.Eur.J.Med.Chem.,2014,81:237-244
[10]Akrami H,Mirjalili B F,Khoobi M,et al.Eur.J.Med.Chem.,2014,84:375-381
[11]Wang C,Wu Z,Cai H,et al.Bioorg.Med.Chem.Lett.,2015,25(22):5212-5216
[12]Hong C,Luo W,Yao D,et al.Bioorg.Med.Chem.,2014,22(12):3213-3219
[13]Giacobini E.Neurochem.Res.,2003,28(3):515-522
[14]Xie S S,Wang X B,Jiang N,et al.Eur.J.Med.Chem.,2015,95:153-165
[15]Alpan A S,Parlar S,Carlino L,et al.Bioorgan.Med.Chem.,2013,21(17):4928-4937
[16]Mohammadi-Khanaposhtani M,Saeedi M,Zafarghandi N S,et al.Eur.J.Med.Chem.,2015,92:799-806
[17]Asadipour A,Alipour M,Jafari M,et al.Eur.J.Med.Chem.,2013,70:623-630
[18]Gao X,Zhou C,Liu H,et al.J.Enzyme Inhib.Med.Chem.,2017,32(1):146-152
[19]Liu H,Liu L,Gao X,et al.Eur.J.Med.Chem.,2017,126:810-822
[20]Gazzard L,Williams K,Chen H F,et al.J.Med.Chem.,2015,58(12):5053-5074
[21]Niu C,XU Y,Xu Y,et al.J.Phys.Chem.B,2005,109(49):23730-23738
[22]Puiatti M,Borioni J L,Vallejo M G,et al.J.Mol.Graph.Model.,2013,44:136-144
[23]Zhu X L,Yu N X,Hao G F,et al.J.Mol.Graph.Model.,2013,41:55-60
[24]Zhou A,Hu J,Wang L,et al.J.Mol.Model.,2015,21(10):277
[25]Shi J,Tu W,Luo M,et al.Mol.Simul.,2016,43(2):102-109
[26]Mohammadi T,Ghayeb Y.J.Biomol.Struct.Dyn.,2017:1-13
[27]Hossain T,Saha A,Mukherjee A.J.Biomol.Struct.Dyn.,2017:1-12
[28]El-Malah A,Gedawy E M,Kassab A E,et al.Arch.Pharm.Chem.Life Sci.,2014,347(2):96-103
[29]Molinuevo J L,Berthier M L,Rami L.Arch.Gerontol.Geriatr.,2011,52(1):18-22
[30]Knowles J.Core Evidence,2006,1(3):195-219
[31]Marco L,Carreiras M C.Recent Pat.CNS Drug.Discovery,2006,1(1):105-111
[32]Wolfson C,Oremus M,Shukla V,et al.Clin.Ther.,2002,24(6):862-886
[33]Bono G F,Simo-Silva D P,Batistela M S,et al.Neurochem.Int.,2015,81:57-62
[34]Kryger G,Silman I,Sussman J L.Structure,1999,7(3):297-307
[35]Larkin M,Blackshields G,Brown N P,et al.Bioinformatics,2007,23:2947-2948
[36]Morris G M,Goodsell D S,Halliday R S,et al.J.Comput.Chem.,1998,19(14):1639-1662
[37]Cornell W D,Cieplak P,Bayly C I,et al.J.Am.Chem.Soc.,1995,117(19):5179-5197
[38]Wang J,Wolf R M,Caldwell J W,et al.J.Comput.Chem.,2004,25(9):1157-1174
[39]Bayly C I,Cieplak P,Cornell W,et al.J.Phys.Chem.,1993,97(40):10269-10280
[40]Jorgensen W L,Chandrasekhar J,Madura J D,et al.J.Chem.Phys.,1983,79(2):926-935
[41]Rycaert J P,Ciccotti G,Berendsen H J C.J.Comput.Phys.,1977,23:327-341
[42]York D M,Darden T A,Pedersen L G.J.Chern.Phys.,1993,99(10):8345-8348
[43]KumariR,KumarR,OpenSource Drug Discovery Consortium,et al.J.Chem.Inf.Model.,2014,54(7):1951-1962
[44]Saíz-Urra L,Cabrera M A,Froeyen M.J.Mol.Graph.Model.,2011,29(5):726-739
[45]El-Barghouthi M I,Jaime C,Al-Sakhen N A,et al.J.Mol.Struct.THEOCHEM,2008,853(1):45-52
[46]Fajer P,Fajer M,Zawrotny M,et al.Methods Enzymol.,2015,563:623-642
[47]Sa R,Fang L,Huang M,et al.J.Phys.Chem.A,2014,118:9113-9119
[48]Jamshidi S,Rafii-Tabar H,Jalili S.Mol.Simul.,2013,40:469-476
[49]Lobanov M Y,Bogatyreva N S,Galzitskaya O V.Mol.Biol.,2008,42(4):623-628
[50]Hu G D,Zhu T,Zhang S L,et al.Eur.J.Med.Chem.,2010,45(1):227-235
[51]Wu E L,Han K L,Zhang J Z H.Chem.Eur.J.,2008,14(28):8704-8714
[52]Laskowski R A,Swindells M B.J.Chem.Inf.Model.,2011,51:2778-2786
Interaction Mechanism Between AChE and 1,7-Diazacarbazole Inhibitors Based on Molecular Dynamics Simulations
ZHAO Teng-TengYANG Xue-Yu DONG Ke-Ke ZHU Xiao-Lei*
(State Key Laboratory of Materials-Oriented Chemical Engineering,College of Chemical Engineering,Nanjing Tech University,Nanjing 210009,China)
The molecular docking,molecular dynamics (MD)simulation,and binding free energy analysis are used to gain the insight into the binding mechanism of three 1,7-diazacarbazole derivatives (marked as M1,M2,and M3,respectively)with AChE at the atom level.The electrostatic and van der Waals (vdW)interactions of the three inhibitors with AChE are analyzed and discussed.The ranking of the computed binding free energies based on MM-PBSA method is consistent with the ranking of experimental bioactivities for the three inhibitors.The hydrogen-bond interactions of the inhibitors with S286 are favorable to the binding affinity of inhibitors to AChE.The van der Waals interactions,especially the key contacts with W279 and Y334 have larger contributions to the binding free energy and play an important role in distinguishing the bioactivities of M1(or M2)and M3.
Alzheimer′s disease;AChE;1,7-diazacarbazole inhibitor;molecular dynamics simulation;MM-PBSA
A
1001-4861(2017)11-2065-10
10.11862/CJIC.2017.250
2017-08-31。收修改稿日期:2017-09-27。
國家自然科學基金(No.21276122)和國家自然科學基金重大研究計劃培育項目(No.91434109)資助。
*通信聯系人。E-mail:xlzhu@njtech.edu.cn
猜你喜歡
天天愛科學(2022年9期)2022-09-15 01:12:54
天天愛科學(2022年4期)2022-05-23 12:41:48
當代水產(2022年3期)2022-04-26 14:26:56
科學大眾(2021年9期)2021-07-16 07:02:54
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
軍事文摘(2020年20期)2020-11-28 11:42:50
航空世界(2020年10期)2020-01-19 14:36:20
中國外匯(2019年17期)2019-11-16 09:31:14
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
