固相萃取-超高效液相-串聯(lián)質譜法檢測水產品中漁藥殘留
2017-11-15 10:52:59張海霞王迎迎
黑龍江水產 2017年5期
關鍵詞:檢測
張海霞 王迎迎
(哈爾濱市農產品質量安全檢驗檢測中心 黑龍江 哈爾濱 150070)
固相萃取-超高效液相-串聯(lián)質譜法檢測水產品中漁藥殘留
張海霞 王迎迎
(哈爾濱市農產品質量安全檢驗檢測中心 黑龍江 哈爾濱 150070)
在水產品養(yǎng)殖過程中,由于使用者對氯霉素、磺胺類、喹諾酮類漁藥及三苯甲烷類染料的毒性和危害認識不足,存在違規(guī)使用的現(xiàn)象.漁藥殘留常見危害為藥物通過較長時間的蓄積,對人體產生的慢性毒性作用.人們長期食用這類含漁藥殘留的水產品后,藥物在人體內蓄積,達到一定濃度后,就會對人體產生各種慢性毒性作用,損害肝臟、腎臟、消化系統(tǒng)、神經系統(tǒng)、造血系統(tǒng)、循環(huán)系統(tǒng)等.農業(yè)部235號公告規(guī)定了其限量標準,農業(yè)部和黑龍江省農監(jiān)局每年要對水產品中這四類漁藥的殘留進行四次例行監(jiān)測,以保證水產品的質量安全.目前,水產品中漁藥殘留的檢測方法主要有酶聯(lián)免疫吸附法、高效液相色譜法、氣相色譜法、氣相色譜-串聯(lián)質譜法、液相色譜-串聯(lián)質譜法、電化學分析、毛細管電泳等,使用的前處理方法主要有液液萃取法、固相萃取法等.這些方法對漁藥殘留的檢測多是按同族藥物或單一藥物來開發(fā),這些方法的不足之處在于僅可以檢測單一或幾個目標化合物,檢測效率低,成本高,造成檢測儀器設備、檢測人員的浪費.本實驗采用Prime HLB固相萃取柱凈化,超高效液相色譜-串聯(lián)質譜法一次性檢測4類12種漁藥殘留,檢測效率高,成本低,適用于越來越繁重的突發(fā)事件和應急監(jiān)測工作.
1 材料與方法
1.1 儀器、試劑與材料
儀器:超高效液相色譜-串聯(lián)四級桿質譜聯(lián)用儀Waters Acquity UPLC-TQD(美國Waters公司),配有電噴霧離子源;Milli-Q高純水發(fā)生器(美國Millipore公司);高速離心機(Sigma);高速組織勻質儀、渦旋振蕩器(IKA);氮吹儀(美國Organomation);pH計、電子天平(Mettler Toledo);超聲波清洗器(KUDOS);料理機(博朗);試劑:優(yōu)級純的乙腈、甲酸、氯化鈉、鹽酸羥胺、對甲苯磺酸、無水乙酸鈉、冰乙酸(科密歐);乙腈、甲醇色譜純(美國Fisher公司);甲酸色譜級(迪馬);Prime HLB固相萃取柱(Waters);標準品均購自Dr.Ehrenstorfer.樣品:供試樣品鯉魚,購自當地超市.魚去鱗,取脊背上的魚肉,切成小塊,在料理機中絞碎.
1.2 樣品的提取
稱取5.0g絞碎的魚肉樣品于50mL具塞離心管內,加入混合內標工作溶液,渦旋混勻.加入1.5mL20%的鹽酸羥胺溶液,2.5mL1.0mol/L的對甲苯磺酸溶液,5.0mL乙酸鹽緩沖溶液,乙腈8mL,渦旋混勻后,超聲5min,再加入8g氯化鈉,渦旋混勻60s,7000r/min離心5min,收集上清液的乙腈層于50mL燒杯中.再向離心管中加入乙腈8mL,渦旋混勻60s,7000r/min離心5min,合并乙腈層的提取液.
1.3 樣品的凈化
Prime HLB固相萃取柱,加入與提取液pH值相近的甲酸乙腈1mL進行固相萃取小柱的活化.將50mL燒杯中的提取液轉移至固相萃取小柱,收集全部流出液,40℃下氮吹至近干.用200μL乙腈溶解后,800μL水定容至1mL,過0.22μm的濾膜.供超高效液相色譜-串聯(lián)質譜測定.
1.4 儀器條件
超高效液相色譜條件:色譜柱: Waters ACQUITY UPLC BEH C18柱(1.7μm,2.1x50 mm );柱溫: 35℃; 進樣體積: 5μL;流速: 0.3mL/min;流動相1:A為乙腈, 流動相B為0.1% 甲酸溶液.具體洗脫程序:0~2.0min10~25%A,2.0~3.0min25~45%A,3.0~4.0min45~90%A, 4.0~5.5min90%A,5.6~7.0min10A.
質譜條件:離子源: 電噴霧電離源( ESI),正、負離子模式;源溫度: 110℃;毛細管電壓:+0.5kV,-2.0 kV;脫溶劑溫度:400℃;脫溶劑氣流速(N2):800L/h;錐孔氣速: 50L/h.采用MRM多反應檢測模式進行檢測.
2 結果與分析
2.1 提取方法
水產品中磺胺類、喹諾酮類和氯霉素的提取試劑主要有乙酸乙酯、甲醇、不同酸度的乙腈等,而孔雀石綠的提取采用了乙腈-鹽溶液或乙腈作為提取試劑[12].本實驗對比了乙腈和乙腈-鹽溶液對魚肉中的12種化合物的提取效率,結果表明,在使用單一的乙腈提取時,孔雀石綠、隱色孔雀石綠及其內標物的提取效率為零,沒有任何回收,其余的化合物的回收較好.采用乙腈-鹽溶液為提取試劑,在保證了磺胺類、喹諾酮類和氯霉素的提取效率的前提下,孔雀石綠和隱色孔雀石綠的回收率能滿足要求.水產品中漁藥殘留的提取主要有高速組織勻質、超聲波提取、震蕩提取,本實驗對以上三種提取方法進行對比.高速組織勻質機進行勻質提取時,由于魚肉較粘,會大部分樣品粘黏在勻質機的刀頭上,降低提取效率.震蕩和超聲波提取都能達到較好的提取效果,但震蕩提取時離心管是橫放在振蕩器上的,提取液容易從離心管蓋溢出,綜合考慮提取效率及可操作性,選取超聲波提取.
2.2 樣品凈化
基質效應是影響LC-MSMS定量分析準確性的重要因素.樣品凈化效果不好會影響化合物的離子化,基質效應強,導致色譜峰峰型不好,并且對色譜柱和質譜有污染.基質效應通過對每個待測物的基質增強或抑制進行評價.具體的計算公式為ME=Rm/Rsⅹ100%,其中Rm為目標化合物在空白基質溶液中的響應值,Rs為目標化合物初始流動相溶液中的響應值[13].固相萃取柱的基本操作步驟為活化、上樣、淋洗、洗脫.Prime HLB固相萃取柱的工作原理是吸附提取液中的雜質已達到凈化的目的,產品的使用方法是不用活化即可使用.為了驗證不活化的固相萃取柱的去除雜質的效果,比對了固相萃取柱不活化、樣品空白提取液活化和pH=1.40的甲酸乙腈(與提取液中的pH值相近)活化三種不同的方式.以基質效應的強弱來衡量不同活化方式的凈化效果.三種不同活化方式的基質效應比較見表1.由表可見,除隱色孔雀石綠為基質減弱效應外,其余參數的三種活化方式均為基質增強效應,其中樣品空白提取液活化和pH=1.40的甲酸乙腈活化的基質效應差異不明顯.使用樣品提取液進行活化,需要每次檢測前都找到陰性樣品,增加工作量和工作難度,最終本實驗選取pH=1.40的甲酸乙腈為活化試劑.
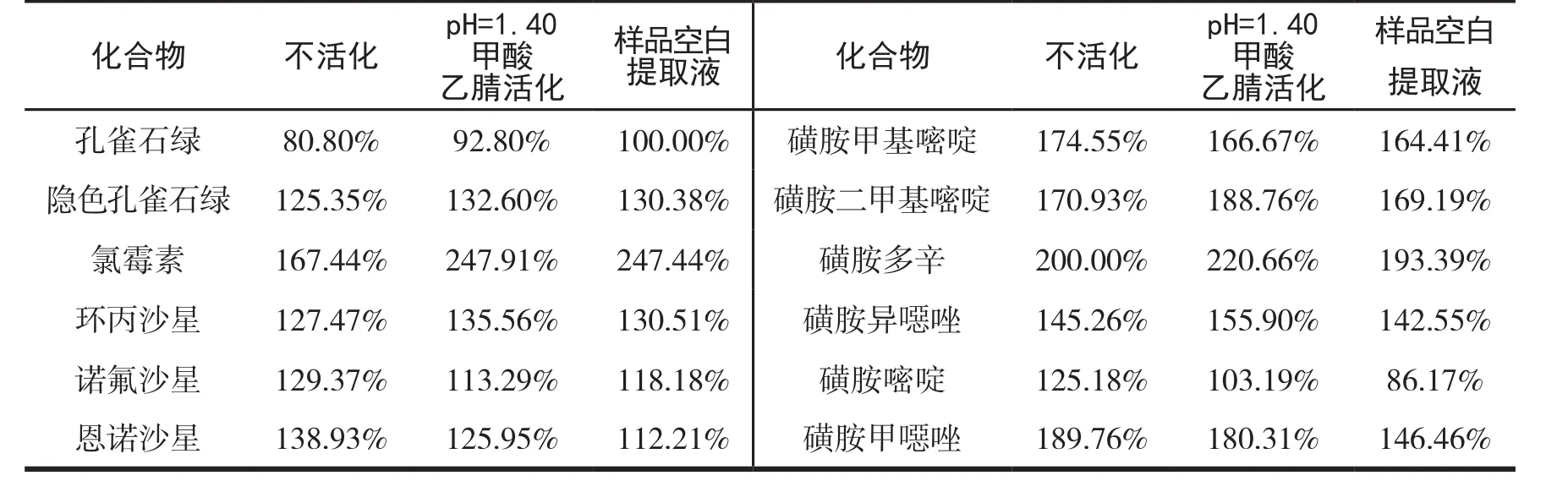
表1 三種不同活化方式的基質效應對比
2.3 質譜方法的優(yōu)化
配制濃度為1mg/kg的12種漁藥及其內標物的標準溶液,以流動注射方式連續(xù)進樣,進行母離子掃描.按照歐盟(2002/657/EC)指令規(guī)定,低分辨率質譜聯(lián)用檢測應確定在母離子的基礎上選擇兩個以上子離子.在確定各個化合物母離子后,對其母離子進行碰撞掃描,每個化合物再選擇2個響應值高的特征離子對作為定量及定性離子,同時對其進行MRM參數優(yōu)化.12種漁藥及其內標化合物中只有氯霉素及其內標物為ESI-模式,其余的參數均為ESI+模式.優(yōu)化的質譜參數見表2.各化合物的總離子流圖見圖1.
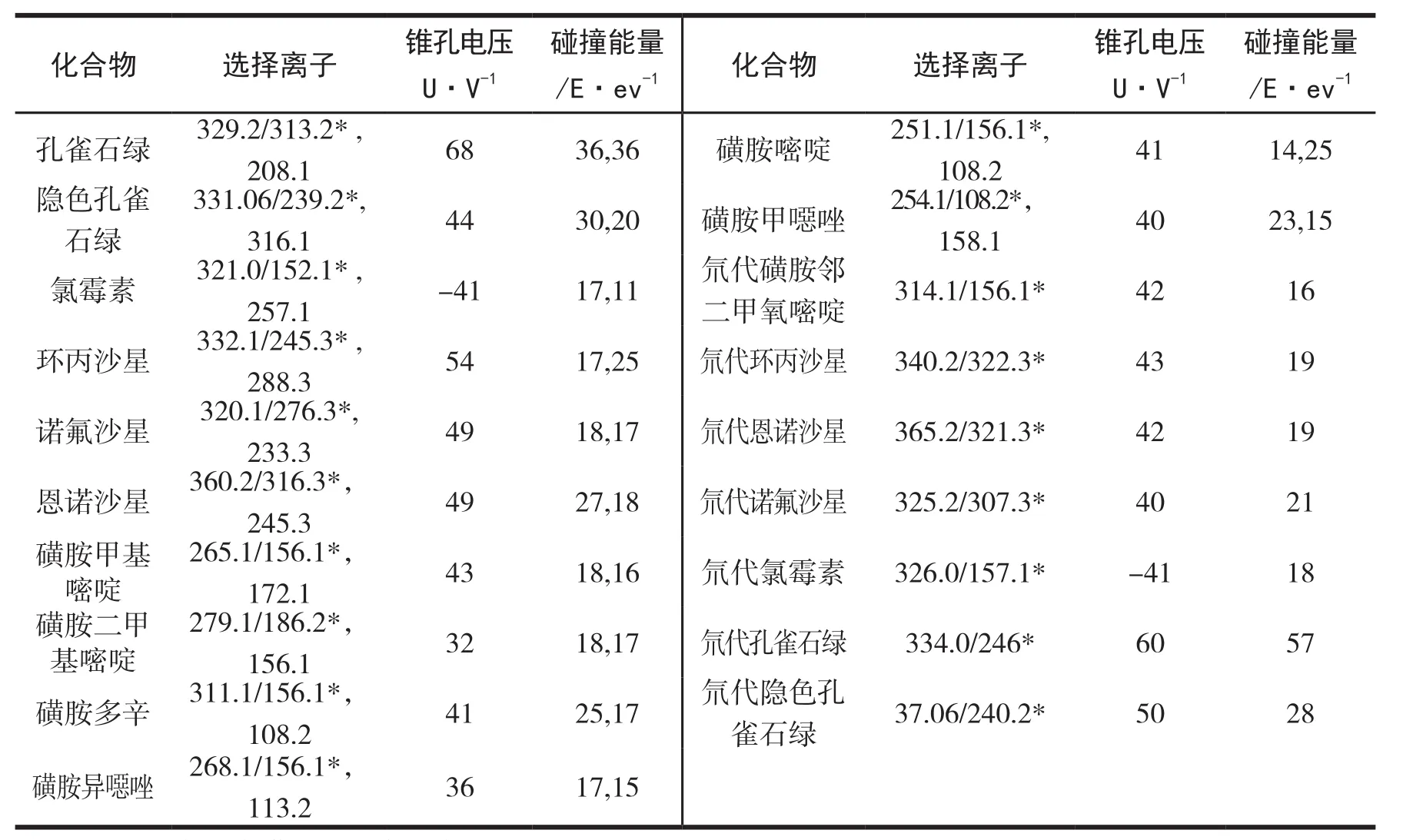
表2 12種漁藥及內標標品多反應監(jiān)測掃描模式(MRM)的質譜參數
圖1 12種化合物及其內標物的總離子流圖
2.4 液相方法的優(yōu)化
由于這12種化合物及其內標物的化學性質差異較大,在質譜中采用了正離子和負離子的采集模式,為了使每個化合物能夠更好的離子化,需要對使用的流動相體系進行優(yōu)化.根據查閱的文獻及本實驗室的工作經驗,對這12種化合物儀器分析時所采用有機相主要有乙腈、甲醇;水相主要有水、甲酸-水、乙酸銨.本實驗進樣適當濃度的混合標準品,對乙腈-5mmol/L乙酸銨、乙腈-0.1%甲酸、甲醇-水三種流動相進行優(yōu)化.各待測物在不同流動相體系中的響應值區(qū)別見圖2.由圖2可以看出,以乙腈-5mmol/L乙酸銨為流動相時氯霉素的響應值高,但喹諾酮化合物的響應值低,其中環(huán)丙沙星沒有響應;以甲醇-水為流動相時磺胺類化合物沒有響應或響應值低;以乙腈-0.1%甲酸為流動相時,氯霉素的響應值較以乙腈-5mmol/L乙酸銨為流動相低,但能滿足檢測需求,其他化合物的響應值均較高,也在檢測需求范圍內.最終選擇以乙腈-0.1%甲酸為流動相.

圖2 待測物在不同流動相體系中的響應值
2.5 方法的線性、檢出限和定量限
為了消除基質效應對定量結果造成的影響,以不含待測物的空白樣品為基質液,配制成基質標準溶液,磺胺類、氯霉素、喹諾酮類化合物濃度分別為10、50、100 、200、300、500、1000μg/mL,孔雀石綠和隱色孔雀石綠的濃度分別為5、10、15 、20、25、50、100μg/mL,進行方法線性的測定.以待檢參數的面積(y)為縱坐標,質量濃度(x)(mg/L)為橫坐標,建立標準曲線,得到線性回歸方程.線性范圍及相關系數見表3.以目標化合物在空白基質中的信噪比(S/N)來獲得檢出限和定量限,S/N=3時對應的含量為檢出限(LOD),S/N=10時對應的含量為定量限(LOQ).

表3 12種漁藥的線性范圍、線性方程及相關系數
2.6 方法的回收率、精密度
按照不同化合物的檢出限和國家規(guī)定的限量標準,每種化合物在樣品中添加低、中、高三種水平濃度的標品,每個水平重復6次,按照2.2的試驗方法進行處理,計算方法的回收率和精密度.具體的添加濃度、回收率和精密度見表4.回收率在70.3-118.7%之間,精密度為1.5-6.7%,符合國家檢測標準的要求.
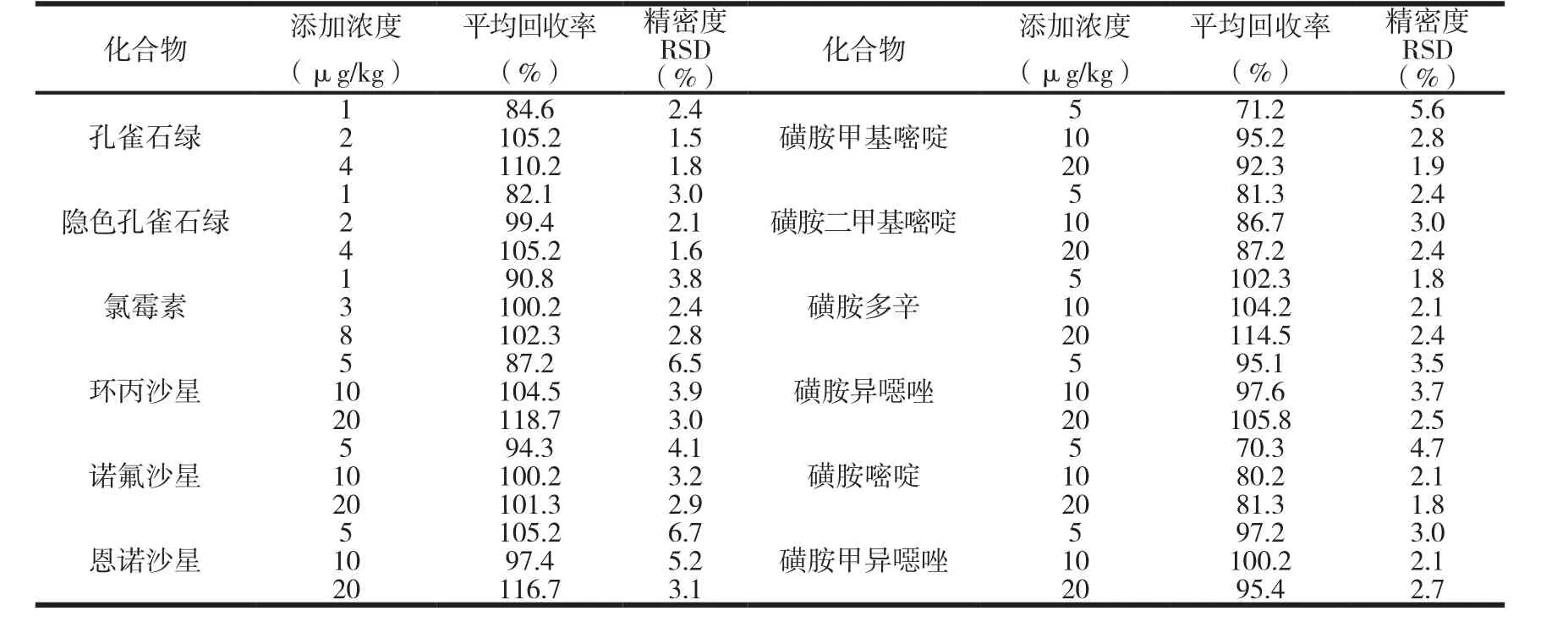
表4 方法的添加回收率和精密度(n=6)
3 結論
本實驗采用乙腈-鹽溶液提取,Prime HLB固相萃取柱凈化,超高效液相色譜-串聯(lián)質譜分析,建立了能一次性檢測水產品中磺胺類、喹諾酮類、孔雀石綠以及氯霉素四類12種漁藥殘留的方法.通過對方法的檢出限、回收率、精密度、線性范圍等方法學進行驗證,該方法滿足對這12種漁藥檢測的要求.與傳統(tǒng)的方法相比,本實驗的方法快速、簡單、靈敏、成本低、效率高,適合實驗室進行批量樣品的檢測.
略)
張海霞 性別:女 職稱:高級農藝師 電話:15204666138 Email:zhanghaixianj@163..com
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
