美國藥品上市后研究的監管制度及其對我國的啟示Δ
2017-11-16 06:36:01羅雪燕賴寒陳紹成李俊重慶第二師范學院生物與化學工程系重慶400067
中國藥房 2017年31期
羅雪燕,賴寒,陳紹成,李俊(重慶第二師范學院生物與化學工程系,重慶400067)
美國藥品上市后研究的監管制度及其對我國的啟示Δ
羅雪燕*,賴寒,陳紹成,李俊#(重慶第二師范學院生物與化學工程系,重慶400067)
目的:為完善我國Ⅳ期臨床試驗的監管提出建議。方法:通過概述美國藥品上市后研究制度,分析美國FDA對藥品上市后研究的監管(包括關鍵要素、監管流程、配套監管系統和強制措施),提出完善我國Ⅳ期臨床試驗監管的建議。結果與結論:美國藥品上市后研究包括上市后承諾研究(PMR)和上市后要求研究(PMC)。監管過程中的關鍵要素包括監管主體(由藥品評價和研究中心下屬的新藥辦公室負責)、關鍵文件(包括幫助FDA和申請人達成研究協議的文件和用于對已確定的研究進行過程跟蹤和監督的文件)、重要時間節點(明確提交相關材料的特定節點日期);監管流程包括制訂研究草案、審核研究報告;美國FDA建立了PMC/PMR數據庫作為配套監管系統,并分別針對PMC和PMR制定了相應的強制措施。我國相關監管部門應轉換監管思路、充分發揮政府的引導和監督作用,加強藥品上市前、后監管的銜接,制訂特色化Ⅳ期臨床方案,建立Ⅳ期臨床試驗數據管理系統,加強全過程監管,借鑒FDA“事前制定計劃,事中動態追蹤,事后依法處理”的監管方式來完善對我國Ⅳ期臨床試驗的監管。
上市后承諾研究;上市后要求研究;監管;Ⅳ期臨床試驗;美國
藥品上市前雖然經歷了嚴格的藥學、非臨床和臨床研究,但藥品上市的決定只是基于已有的非臨床和臨床數據進行風險評估之后的平衡決策結果,作為決策基礎的試驗數據也有其固有缺陷[1-2]。因此,與我國一樣,美國FDA在批準新藥上市的同時,或者在批準藥品上市后,只要認為有必要獲得更多有關藥品風險、利益及最佳使用方法等信息資料,就可以依照法規授權,建議或要求申請人進行藥品上市后研究[3]。而對于這類“研究”,美國《食品藥品管理補充法案》(2007年)將其區分為“臨床試驗(Clinical trials)”和“研究(Study)”兩類[4],并最終將其統一界定為藥品的“上市后研究和臨床試驗”(Post-marketing clinical trial and study,以下簡稱“上市后研究”)制度[5]。
筆者擬通過文獻研究,從藥品監管者的角度出發,圍繞監管中的關鍵要素、監管流程、配套監管系統和強制措施四個方面,系統分析美國FDA對藥品上市后研究的監管方式。在此基礎上,借鑒美國FDA的監管經驗,對完善我國Ⅳ期臨床試驗監管制度提出建議。
1 美國藥品上市后研究制度概述
美國藥品上市后研究制度是指針對上市后藥品的安全性和有效性研究,包括臨床試驗、調查、動物研究以及實驗室研究等。具體來說,即根據醫藥學最新學術水平,從藥理學、藥劑學、臨床醫學、藥物流行病學及藥物政策等方面,對已批準的藥品在社會人群中的療效、不良反應、用藥方案、穩定性、費用等多方面是否符合安全、有效、經濟合理等原則作出科學的評價[6]。根據研究是否具有法律強制性,藥品上市后研究可分為上市后承諾研究(Post-marketing commitment research,PMC)和上市后要求研究(Post-marketing requirement research,PMR)。
1.1 PMC
根據美國國會1997年通過的《1997年食品藥品管理現代化法》(Food and Drug Administration Moderization Act of 1997,FDCA)”,以及2002年通過的《2002年公共健康安全和生物恐怖預案應對法》中對FDCA 506B條款有關藥品上市后研究的補充,規定藥品PMC,即藥品上市后需要開展的非法定的、未經FDA強制要求的相關研究屬協議任務。研究內容一般由FDA提出,需要上市藥品責任人同意并承諾進行研究,該行為不受法律約束,申報者僅需每年向FDA報告研究的實施進程[7]。
PMC的目的在于對潛在風險較大的藥品的風險、效益比進行持續、動態評估,以更好地保障患者用藥安全。其研究內容相對寬泛,多是為了獲得更多藥品風險、效益及最佳使用信息,解決上市使用中出現的安全性問題和顧慮,包括對藥物的疾病史與不良反應發生率的流行病學研究,進一步確定療效的臨床研究和進一步的質量研究等[8]。
1.2 PMR
2007年,美國簽署并發布了《食品藥品監督管理局2007修正法案》(Food and Drug Administration Amendments Act of 2007,FDAAA)[9],其第9部分“加強對藥品上市后的安全監管”中的505(o)條授權FDA,允許FDA在處方新藥或新生物制品上市批準的同時,或在批準上市后,當意識到藥品的新安全性信息或對藥品安全性存在威脅時,可以依法要求上市藥品責任人進行該藥物或生物制品的上市后研究。
上述條款賦予了FDA更多用于藥品上市后安全監管和風險管理的權力和資金,標志著FDA的這一權力和職能正式獲得法律認可。然而,并非所有藥品都有開展PMR的義務,僅有處方藥和專利藥會受到505(o)條款的約束。而且通常來說,PMR主要針對已經發現風險或有嚴重風險信號的產品。因而FDA在開展一項PMR之前,必須明確藥物相關的不良反應/事件報告,并確認現有藥物警戒系統不能達到下述目的:(1)評估上市藥品有關的已知嚴重風險;(2)評估上市藥品有關的嚴重風險信號;(3)識別已知數據所提示的潛在嚴重風險[10]。基于上述原則,FDAAA授予FDA權力在以下幾種情況下,可以向申請人提出PMR:(1)加快程序審批的藥品;(2)依據動物藥效學研究結果審批的藥品;(3)兒童用藥的延遲研究;(4)新批準的處方新藥及新生物制品的風險評價。
1.3 PMC和PMR的實施情況
一個藥品可能擁有多個PMR和PMC,但一般來說,PMR的數量要多于PMC。據FDA官方統計,每年新備案的PMR數量大約是PMC的3~5倍。而根據FDA官方網站最新發布的《藥品和生物制品開展上市后要求和承諾情況的報告》統計,截至2015年9月,共有269個申請人提交的856個新藥和新生物制品的上市后研究項目(包括PMR和PMC)記錄在案,其中包括194個申請人提交的716個新藥申請(New drug application,NDA)、74個申請人提交的140個生物制劑許可申請(Biologic license application,BLA)項目。
如上文所述,美國FDA對PMR和PMC的約束力有所不同,在一定程度上導致兩者在開展過程中的依從情況存在一定差異。總體來看,雖然FDA記錄的PMR項目數量要遠高于PMC,監管難度也較PMC更大,但PMR整體的依從水平要高于PMC,主要表現在研究按預定進度開展的比例相對較高、延期比例較低,詳見表1(數據截至2015年9月)。

表1 PMR和PMC的開展情況(%%)Tab1 The progress of PMR and PMC(%%)
2 美國藥品上市后研究的監管
如上文所述,美國藥品上市后研究可分為PMR和PMC兩種。但總體來看,FDA對PMR和PMC的監管方式基本一致,所涉及的監管要素、流程和職責分配存在較多重合和相似之處。鑒于此,下文分析美國藥品上市后研究的監管方式時,將不針對PMR和PMC作分別闡述,僅在兩者存在差異時予以區分。
2.1 監管過程中的關鍵要素
2.1.1 監管主體在美國上市銷售的藥品,其上市后研究的監管主要由藥品評價和研究中心(Center for Drug Evaluation and Research,CDER)下屬的新藥辦公室(Office of new drug,OND)負責,監測則由流行病學辦公室(Office of Surveillance and Epidemiology,OSE)輔助完成[11]。具體來說,一般由來自OND新藥評審團隊的專業評審員和管理人員完成具體監督和項目評價工作,而這些人員依照其職責設置,可以分為開展具體工作的PMR/PMC的制訂協調員、評審員、項目管理員、跟蹤協調員、質量管理員等,以及負責整體協調和管理的項目管理經理、評審管理員[12]。
2.1.2 關鍵文件在藥品上市后研究方案制訂和開展的過程中,FDA和申請人之間所涉及的關鍵文件按照其作用可分為兩類:一類是幫助FDA和申請人達成研究協議的文件,包括FDA完成審評后起草的“PMR/PMC草案”和最終發送給申請人的“PMR/PMC通知件”;另一類是用于對已確定的研究進行過程跟蹤和監督的文件,一般由申請人按規定時間向FDA提交,包括研究的“年度狀態報告”“最終報告”和“完善PMR/PMC的補充申請文件”等。
2.1.3 重要時間節點為了跟蹤某藥品上市后研究的完成進度,并監督申請人在規定時間內完成相關研究工作,FDA通常會要求申請人在研究進度計劃和研究申請信中明確提交相關材料的特定節點日期,并將其作為PMR/PMC協議內容的一部分。這些時間節點可以為某一個具體時間,如最終研究協議提交日期、最終協議提交日期或最終研究報告提交日期;也可以為“距前一個節點的時段”,如“最終協議提交日期后的第n個月完成研究”“研究實際完成后的第n個月內提交最終研究報告”等。
2.2 監管流程
從美國藥品上市后研究的定義和基本要求可知,FDA的相關監管工作主要可分為兩個部分:(1)藥品上市前,與申請者協定研究內容,制訂研究草案;(2)藥品上市后,監督申請者按計劃提交研究報告,審核其是否達到要求。
2.2.1 制訂研究草案PMR或PMC研究草案的制訂工作一般在NDA審評基本完成后開始,至FDA向申請者發送審批件(Approval letter)時正式結束。這一階段相關監管人員的主要職責為:①明確藥品是否有必要開展PMR或PMC;②制訂PMR或PMC研究草案,通過溝通交流與申請人就研究內容和進度安排達成一致;③將最終協定的研究方案告知申請人。監管中的方案制訂流程見圖1。
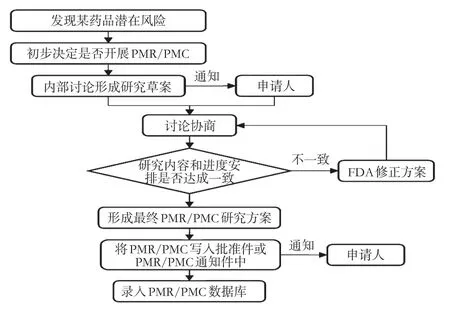
圖1 FDA對藥品上市后研究監管中的方案制訂流程Fig1 Protocol development flow of FDA’s supervision on drug post-marketing research
2.2.2 審核研究報告申請人按協議研究方案開展藥品上市后研究后,需要按研究進度計劃定期向FDA提交研究報告。在這個過程中,FDA相關監管人員的主要職責在于:①跟蹤研究過程,督促申請人按期提交研究報告;②審核研究報告的真實性和科學性,及時發現問題并要求申請人補充材料;③確認已有研究是否實現了證實藥品安全性和有效性的目的。監管中的報告審核流程見圖2。
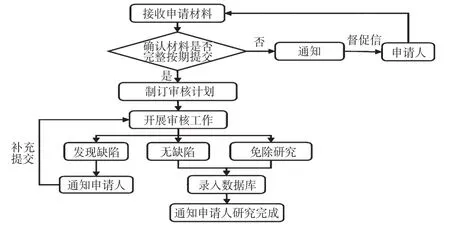
圖2 FDA對藥品上市后研究監管中的報告審核流程Fig2 Report review flow of FDA’s supervision on drug post-marketing research
2.3 配套監管系統
如上文所述,FDA與申請者達成一致的藥品上市后研究協議后,PMR/PMC跟蹤協調員會將藥品的基本信息和最終確定的研究內容登記到PMC/PMR數據庫中,并在未來的跟蹤和監督過程中,以數據庫記錄的研究報告提交情況為依據,確定研究的進展狀況。
PMR/PMC數據庫是一個對外公開的數據庫,公眾和企業可以通過檢索此數據庫,了解FDA批準的每個藥品的PMR、PMC的基本信息和實施情況,包括申請者信息、產品信息、PMR和PMC內容描述、年度研究報告提交情況以及“當前狀態”等。其中,“當前狀態”一欄是FDA判定某藥品上市后研究實施情況的重要依據,只有達到“已完成”或“已免除”狀態,才表明該藥品的上市后研究工作全部完成。此外,FDA對PMR/PMC數據庫實施動態管理,數據庫內公布的數據會在每年的1月、4月和10月定期更新。PMR/PMC數據庫中的7種“當前狀態”見表2[13]。

表2 PMR/PMC數據庫中的7種“當前狀態”Tab2 7 kinds of“current condition”in PMR/PMC database
2.4 強制措施
對于PMR,由于其本身具有FDAAA法案賦予的義務性,因而申請者未按要求完成相關研究將被視為違法行為,并會因此受到法律處罰。FDAAA 505(p)條款授權FDA,除非申請人有能夠說服FDA監管人員的充分理由,否則可以依照法律對其采取以下幾種處罰措施[14]:(1)限制相關藥品的州際間貿易;(2)指控相關藥品的標簽錯誤;(3)民事罰款。
而對于PMC,雖然此類研究不屬于法定義務,未在預定時限內按方案完成研究的申請者不會依法受到行政處罰或民事罰款。但對于此類情形,《美國聯邦行政法典》CFR21(Code of Federal Regulations)第9章第356b(e)條提出,FDA局長可以要求申請者通知執業醫師,該藥廠生產、銷售的藥品未能完成上市后研究,以至于可能無法解釋該藥品的某些臨床獲益情況和安全性問題。FDA將在官方網站上發布聲明,告知公眾某藥品的申請者未按要求完成上市后研究,并說明未達到協議要求的、未完成研究的原因。
3 美國上市后研究制度對我國的啟示和建議
通過研究發現,FDA依靠完備、明確的法律條款,充分發揮了自身對藥品上市申請者的指導作用和監管職能,實現了對藥品上市后研究的科學、規范監管。在此基礎上,還通過專有數據庫實現了對藥品上市后研究狀態的實時追蹤,督促企業按照研究計劃完成研究[15]。這對于完善我國藥品Ⅳ期臨床制度,充分發揮Ⅳ期臨床試驗在提高藥品安全性、有效性方面的作用,具有一定啟示和借鑒意義。
3.1 轉換監管思路,充分發揮政府的引導和監督作用
在藥品監管方面,我國國家食品藥品監督管理總局(CFDA)與FDA的監管理念和監管方式存在較大差異,藥品上市后的安全監管也不例外。從美國藥品上市后研究的監管流程可以看出,FDA無論在上市后研究協議的制訂還是執行過程中,都會邀請申請人參與其中,并注重與申請人的溝通交流,從而保證上市后研究設計良好、執行嚴格。
對此,筆者認為,監管者應當意識到企業是產品的第一責任人,政府在藥品監管過程中所扮演的應是引導者和監督者的角色,而非包攬所有責任。因而,在藥品Ⅳ期臨床監管制度的制度設計中,應充分考慮監管部門和申請者之間的責任分配。具體來說,即建議藥品監管部門允許申請者參與試驗內容和試驗進度的制訂,同時,對申請者提出的科學、合理的修正意見予以考慮和采納。而當藥品上市后,CFDA負有告知申請者的主動報告義務,并以此為紐帶保持審評團隊與申請人之間的聯系,持續指導和監控Ⅳ期臨床研究的開展。
3.2 加強藥品上市前后監管的銜接,制訂特色化Ⅳ期臨床方案
無論是上市后的PMC還是PMR,FDA都是基于NDA審評情況,對每個藥品制訂特色化上市后研究方案,并允許申請人根據自身情況與FDA共同修正。這種做法相比于我國依照藥品注冊分類,以“一刀切”的方式管理藥品Ⅳ期臨床試驗更加科學、有效。我國應借鑒美國對PMR和PMC的分類管理,充分考慮藥品本身的風險,以及申請者提交的NDA材料中關于藥品安全性和有效性的證據,以此為依據,有針對性地要求或建議企業開展Ⅳ期臨床試驗,并制訂內容明確、進度安排合理的研究方案。
3.3 建立Ⅳ期臨床試驗數據管理系統,加強全過程監管
數據管理系統是現代化管理的重要工具,高效率、高質量的管理離不開該工具。Ⅳ期臨床試驗周期較長、過程復雜,構建相應的數據庫及數據管理系統對實施高效跟蹤和管理非常必要。借鑒美國PMR/PMC數據庫的管理模式,我國CFDA可建立類似平臺,對企業每年遞交的臨床Ⅳ期臨床試驗報告進行存儲,并跟蹤發布各品種藥品臨床Ⅳ期進展及結果的相關信息,以實時關注藥品臨床Ⅳ期的實施情況。基于該數據庫的跟蹤監測情況,CFDA可每年匯總并撰寫概況報告,總結年度Ⅳ期臨床試驗的安全情況;同時,數據庫中記錄的時間節點能夠幫助監管部門及時發現、糾正未按規定開展Ⅳ期臨床試驗的情況,提高藥品上市后安全監管的效果。
4 討論
藥品上市所依據的藥學、非臨床和臨床研究往往存在一定固有缺陷。因此對于大多數藥品,尤其是首次上市的新藥來說,上市后繼續在更廣泛的人群中開展藥品安全性、有效性研究往往是必要的。我國長期以來“重審批、輕監管”,因而對藥品上市后研究一直缺乏關注和管理。然而隨著藥品監管部門逐漸重視“過程監管”,以及相關機構改革和制度改革的同步開展,我國應借鑒美國對藥品上市后研究的這種“事前制訂計劃,事中動態追蹤,事后依法處理”的監管方式,以及將藥品監管貫穿藥品上市后整個周期的思路,為我國完善Ⅳ期臨床試驗制度和加強藥品上市后的風險管理提供重要參考。
[1] 趙建中,謝松梅,楊進波,等.不同國家藥品上市后研究管理現狀比較[J].中國新藥雜志,2014,23(22):2589-2592.
[2] 向秋靜,葉樺.關于國外開展藥品上市后再評價相關制度的分析[J].中國藥事,2016,30(4):406-410.
[3] 劉璐,溫寶書,黃清竹,等.Ⅳ期臨床試驗管理體制的研究探討[J].中國新藥雜志,2010,19(17):1503-1507.
[4] 董鐸,孫利華,王丹.美國FDA關于企業開展藥品上市后研究和臨床試驗指南[J].中國新藥雜志,2011,20(9):960-962.
[5] FDA.Food and drug administration amendment act[EB/OL].[2016-11-15].https://www.fda.gov/regulatoryinfor-mation/lawsenforcedbyfda/significantam end ments to the fdcact/food and drugadministratio nam end mentsact of2007/default.htm.
[6] 劉平羽.國外藥品上市后再評價制度簡介[J].上海醫藥,2004,25(5):208-210.
[7] 孫新欣.美國新藥上市后定期匯總報告的研究[J].上海醫藥,2013,34(21):44-47.
[8] 任經天,吳曄,程魯榕.美國藥品上市后研究承諾簡介[J].藥物流行病學雜志,2005,14(2):97-100.
[9] 奚曉云,李國芬.美國、歐盟和日本藥物警戒法規體系簡介[J].藥物流行病學雜志,2010,19(10):587-591.
[10] 郭曉昕,杜曉曦.藥品風險信號的發現與上市后研究[J].中國臨床藥理學雜志,2011,27(8):634-641.
[11] 董鐸,劉翠麗.美國藥品生產企業上市后監測制度研究及啟示[J].中國藥物警戒,2013,10(8):456-459、463.
[12] FDA.Procedures and responsibilities for developing post market in grequirement sandcom mitments[EB/OL].[2016-11-10].http://101.96.10.43/www.fda.gov/downloads/abooutfda/centers offices/office of medical product sandtobacco/cder/manual of policies procedures/ucm120877.
[13] FDA.Postmarketing requirements and commitments:frequently asked questions(FAQ)[EB/OL].[2016-11-08].http://www.fda.gov/drugs/guidance compliancere gulatory in for mation/post-market in gpha seivco mmitments/ucm-070766.htm.
[14] FDA.Notice to industry:post marketing requirements post marketing studies and clinical trials[EB/OL].[2016-11-08].http://www.fda.gov/drugs/guidance compliancere gulatory in for mation/ucm292758.htm.
[15] 單愛蓮,蔣玉鳳.新藥Ⅳ期臨床試驗與藥品上市后再評價的異同點以及存在的問題[J].中國臨床藥理學雜志,2014,30(5):387-390、396.
Supervision System on Drug Post-marketing Research in America and Its Enlightenments to China
LUO Xueyan,LAI Han,CHEN Shaocheng,LI Jun(Dept.of Biology and Chemical Engineering,Chongqing University of Education,Chongqing 400067,China)
OBJECTIVE:To put forward suggestions for improving the supervision of phaseⅣclinical trials in China.METHODS:According to summarizing the post-marketing research in America,FDA’s supervision(including key elements,supervision flow,auxiliary supervision system and enforcement measures)for drug post-marketing research in America was analyzed,and suggestions for the supervision of phaseⅣclinical trials in China was put forward.RESULTS&CONCLUSIONS:The drug post-marketing research in America included post-marketing commitment research(PMR)and post-marketing requirement research(PMC).The key elements included supervision subjects(dealt by Office of New Drugs affiliated to Drug Evaluation and Research Center),key document(including the documents helping FDA and applicants reached a research agreement,and documents for process tracking and supervision in identified studies)and important time node.The supervision flow included developing drafts and reviewing reports.FDA had established PMC/PMR database,which was used as auxiliary supervision system,and relevant enforcement measures were respectively developed for PMC and PMR.Relevant supervision departments in China should converse the supervision ideas,give full play to the government’s guidance and supervision,enhance the connection of supervision between pre-and post-marketing,specially develop phaseⅣclinical program,establish system for phaseⅣclinical trial data,enhance whole process supervision,draw lessons from“pre-process plan,dynamic tracking in the process,and post-process decision according to law”of FDA to improve the supervision of phaseⅣclinical trials in China.
Post-marketing commitment research;Post-marketing requirement research;Supervision;PhaseⅣclinical trials;America
R951
A
1001-0408(2017)31-4330-05
DOI 10.6039/j.issn.1001-0408.2017.31.03
重慶市教委2015年度科學技術研究項目:基于“三醫聯動”模式下的藥品采購制度研究——以重慶市為例(No.KJ1501417);重慶第二師范學院2015年度校級科研項目:基本醫療保險藥品價格談判機制研究——以重慶市為例(No.KY201511B)
*講師。研究方向:藥品政策法規、醫療保障政策。電話:023-86388609。E-mail:375597241@qq.com
#通信作者:講師。研究方向:藥品流通領域政策法規。電話:023-86388609。E-mail:2474912362@qq.com
2017-01-20
2017-08-14)
(編輯:劉明偉)
猜你喜歡
車主之友(2022年6期)2023-01-30 08:01:04
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
車主之友(2022年4期)2022-11-25 07:27:30
車主之友(2022年4期)2022-08-27 00:57:48
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
中國衛生(2016年5期)2016-11-12 13:25:28
中國衛生(2015年5期)2015-11-08 12:09:48
