苝四甲酸二酐薄膜電子結構的同步輻射共振光電子能譜研究?
2017-12-05 02:35:24李智浩曹亮郭玉獻
物理學報 2017年22期
關鍵詞:信號
李智浩 曹亮 郭玉獻
1)(安徽建筑大學材料與化學工程學院,合肥 230601)2)(中國科學院強磁場科學中心,安徽省極端條件凝聚態物理重點實驗室,合肥 230031)
苝四甲酸二酐薄膜電子結構的同步輻射共振光電子能譜研究?
李智浩1)2)曹亮2)?郭玉獻1)?
1)(安徽建筑大學材料與化學工程學院,合肥 230601)2)(中國科學院強磁場科學中心,安徽省極端條件凝聚態物理重點實驗室,合肥 230031)
(2017年5月31日收到;2017年8月23日收到修改稿)
利用基于同步輻射的近邊X射線吸收精細結構譜(NEXAFS)和共振光電子譜(RPES)研究了苝四甲酸二酐分子(PTCDA)薄膜的電子結構.碳K邊NEXAFS譜中能量小于290 eV的四個峰對應于PTCDA分子不同化學環境碳原子1s電子到未占據分子軌道的共振躍遷.RPES譜中觀察到共振光電子發射和共振俄歇電子發射導致的共振峰結構,以及二次諧波激發的碳1s信號.根據電子動能對入射光能量的依賴性分別對三類峰結構進行了歸屬.同時,發現PTCDA分子軌道共振光電子峰的強度具有光子能量依賴性.這種能量選擇性共振增強效應是由于PTCDA分子軌道空間分布差異導致的.共振俄歇峰主要源于高結合能(>4.1 eV)分子軌道能級電子參與的退激發過程.明確RPES實驗譜圖中各個峰結構的起源有助于準確利用基于RPES的芯能級空穴時鐘譜技術定量估算有機分子/電極異質界面處電子從分子未占據軌道到電極導帶的超快轉移時間.
有機半導體,近邊吸收精細結構譜,共振俄歇,同步輻射光電子能譜
1 引 言
有機半導體材料具有優異的光電性質,同時兼具功能多樣性、易加工和價格低廉等優點[1],在有機場效應管(OFET)[2,3]、有機發光二極管(OLED)[4]和有機光伏太陽能電池(OPVC)等[5,6]電子學器件中有著潛在的應用前景[7].在這些器件中,有機半導體/電極異質界面的物理性質是決定器件性能的關鍵性因素之一[8,9].對于太陽能電池材料,匹配的界面能級排列結構有利于載流子的傳輸和收集,對提高電池的轉化效率起決定性作用.界面處超快電子轉移能有效地提高激子解離概率,降低激子復合引起的能量損失,進而提高光電轉化效率.然而,界面處的電荷轉移時間一般在飛秒或亞飛秒量級[10],對測量方法的時間分辨率是一個嚴峻的挑戰.基于同步輻射的芯能級空穴時鐘(core-hole clock,CHC)譜具有超高的時間分辨率,可用于研究原子或分子與電極異質界面處超快電荷轉移動力學.其基本原理見參考文獻[11—13].該技術以能量為探針,以激發態芯能級空穴的壽命(飛秒量級)作為時間參考,通過對比退激發各過程引起的共振峰強度的變化,可獲得亞飛秒甚至阿秒量級的時間分辨率.
苝四甲酸二酐(PTCDA)分子是一種典型的平面共軛結構的有機半導體材料[14,15],帶隙為2.2 eV.它具有一個苝核心和兩個酸酐官能團,分子結構如圖1所示.由于良好的光電性能和化學穩定性,PTCDA分子在有機電子學器件中有著廣泛的應用[16].因此,PTCDA是有機半導體基礎研究的模型分子[17?19].我們利用CHC技術研究PTCDA/TiO2界面電子轉移動力學,通過對比界面處以及薄膜內分子共振光電發射信號積分強度的變化,定量估算電子從最低未占據分子軌道(LUMOs)到TiO2導帶的飛秒(8—20 fs)量級轉移時間[10].然而,計算電子轉移時間時,需排除共振俄歇退激發信號和非共振發射信號對共振光電發射信號積分強度的影響[11,13].積分信號過度采集將導致計算結果的偏離.因此,研究有機分子的電子結構、明確有機分子各退激發過程、提取有效信號的強度,將有助于精確估算界面處超快電子轉移時間.
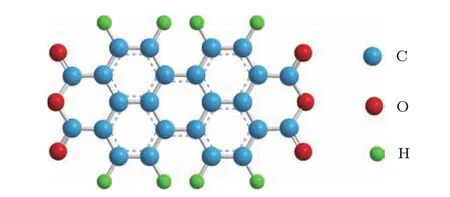
圖1 (網刊彩色)PTCDA分子結構示意圖Fig.1.(color online)Molecular structure of a PTCDA molecule.
圖2為芯能級光激發和退激發過程示意圖.圖2(a)和圖2(b)分別是典型的價電子和芯能級電子光電離過程.終態含有一個單位正電荷.當芯能級電子被電離后,同時會發生俄歇退激發過程(見圖2(c)).價電子填充芯能級空穴,并釋放出能量激發另一個價電子躍遷到真空能級(Evac)之外.圖2(d)是光子吸收過程:芯能級的電子受激躍遷到LUMO軌道.終態是中性的,不帶電.對于碳原子而言,其芯能級空穴的壽命是6 fs[20].這種亞穩定的激發態導致兩種自電離退激發過程.兩者的區別在于LUMO上的受激電子是否參與退激發.圖2(e)中受激電子退激發填充芯能級空穴,同時價電子激發躍遷到Evac之外.此過程被稱為參與退激發(participator decay).其終態帶有一個單位正電荷,與價電子激發終態(圖2(a))相似,出射電子的結合能為常數.由于參與退激發過程增大了電離截面,又稱為共振光電退激發;圖2(f)中受激電子沒有參與退激發過程.價帶上的電子填充了芯能級空穴,同時另一個價電子躍遷到Evac之外.因此,該過程稱之為非參與退激發(spectator decay).這個過程終態類似于俄歇退激發過程(圖2(c)).其出射電子的動能(EK)為常數,故而又稱之為共振俄歇退激發.它的終態含有一個單位的正電荷,由于受到LUMO軌道受激電子的屏蔽作用,共振俄歇電子比常規俄歇電子的動能略大.
本文旨在闡明如何在實驗測量的譜圖中分辨出上述過程導致的峰結構.明確各個峰的起源有助于提取有效的信號用于超快電荷動力學研究.本文使用有機分子束沉積(OMBD)技術制備了苝四甲酸二酐薄膜,利用基于同步輻射的近邊吸收精細結構譜(NEXAFS)和共振光電子譜(RPES)對PTCDA分子的電子結構進行原位測量.分別對NEXAFS測量的碳原子K殼層電子共振躍遷峰和RPES獲得的光電發射峰結構進行了歸屬;解釋光電發射峰及共振增強效應的光子能量依賴性的起源.
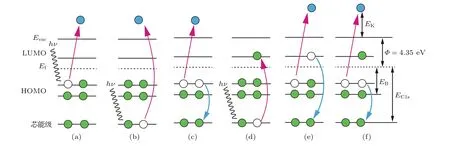
圖2 (網刊彩色)芯能級光激發和退激發過程示意圖Fig.2.(color online)Schematic view of photo-excitation and decay processes.
2 實 驗
通過氬離子刻蝕和真空退火方法得到清潔的金襯底表面.使用光電子能譜(PES)對表面的清潔性進行測量.PTCDA分子在真空中充分除氣,用OMBD的方法沉積到清潔的襯底表面.蒸發速率(約為6.6 ?/min)預先由石英晶體振動膜厚儀測量;實際沉積厚度根據金4f光電子譜強度的衰減進行估算.
NEXAFS和RPES譜是在新加坡光源SINS光束線完成.SINS光束線分析室真空好于1×10?10mbar,配備R4000電子能量分析器.所有的譜圖在室溫下測量.NEXAFS采用全電子產額模式測量.RPES譜測量時使用的光子能量覆蓋碳1s→π*吸收邊.NEXAFS和RPES譜圖強度均對光電流進行歸一化.光子能量使用清潔的Au薄膜4f7/2的結合能(84 eV)進行矯正.譜圖結合能以費米能級為零點.
3 結果與討論
3.1 NEXAFS譜學表征
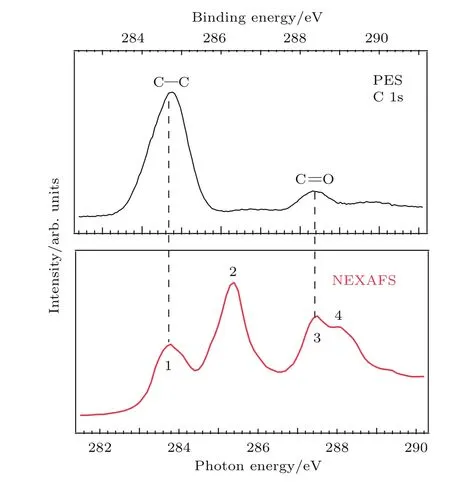
圖3 (網刊彩色)PTCDA薄膜碳1s芯能級PES譜和碳K-邊NEXAFS譜Fig.3.(color online)C 1s core-level spectrum and C K-edge NEXAFS spectrum of a PTCDA thin film.
NEXAFS可以測量電子從芯能級到非占據分子軌道的共振躍遷.圖3為PTCDA薄膜(約33 ?)的碳1s芯能級PES譜及碳原子K邊NEXAFS譜.PES譜激發光子能量為345 eV,電子垂直出射.其中最強峰(約284.8 eV)對應于苝環中碳原子的激發.約288.4 eV的峰對應于PTCDA分子中羰基(C=O)碳原子的激發.高結合能肩峰為震激峰.NEXAFS譜中在283—289 eV范圍內出現的四個尖峰(1—4)分別對應于不同化學環境的碳1s芯能級電子到π*軌道的共振躍遷[21].峰1(約283.8 eV)對應于苝環碳1s電子到LUMO的躍遷;峰2(約285.4 eV)的強度最強,對應于苝環碳原子1s芯能級電子到LUMO+1—LUMO+3的躍遷;相似地,峰3(約287.5 eV)和峰4(約288.2 eV)分別對應于羧基碳1s電子到LUMO和LUMO+1—LUMO+3的躍遷[22,23].
3.2 RPES譜學表征
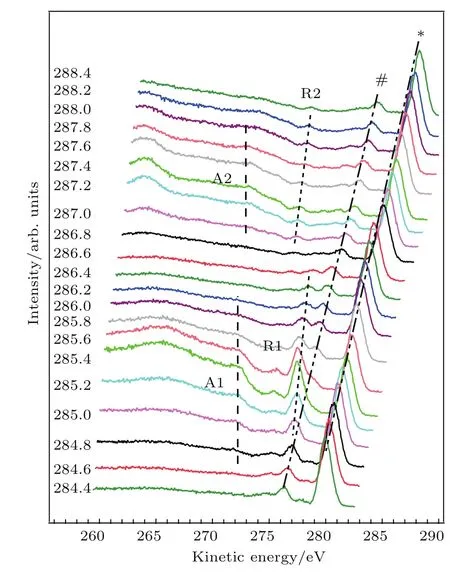
圖4 (網刊彩色)掃描吸收邊能量測量的PTCDA薄膜RPES譜Fig.4.(color online)RPES spectra of a PTCDA thin film measured with photon energy across the C 1s→ π*absorption threshold.
為了明確退激發過程,我們選擇吸收邊能量作為激發光源,掃描價帶附近出射電子動能,獲得RPES譜.如圖4所示,高動能處的*峰和#峰的動能變化是掃描能量變化的兩倍,且峰形與PES結果一致(見圖3碳1s譜).因此,主要來源于二次諧波激發的碳1s芯能級信號.動能小于274 eV的信號的動能不隨光子能量的變化而變化,因此,主要來自于俄歇電子(包括常規俄歇電子和共振俄歇電子)的貢獻.虛線分別標記出了A1,A2,R1,R2共振峰動能隨光子能量的變化.A1峰和A2峰的動能不隨光子能量的變化而變化,屬于共振俄歇退激發(見圖2(f)).R1峰和R2峰的動能變化與掃描能量變化一致,來源于共振光電退激發(見圖2(e)).R1和R2共振峰的強度具有明顯的光子能量依賴性:當能量為248.6—286.4 eV時,可以觀察到明顯的R1共振增強峰;當能量為287.0—288.4 eV時,只觀察到R2共振增強峰[10,22].這種選擇性共振增強效應是由于PTCDA分子軌道空間分布導致的[24]:具有一定能量的光子恰好可以激發芯能級電子躍遷到與價帶分子軌道空間重疊的非占據分子軌道上時,價帶軌道共振增強效應最強.
為了清晰地說明共振增強效應的光子能量依賴性,將285.4和287.6 eV測量的譜圖的動能轉化為結合能,并與非共振能量(60.0 eV)下測量的價帶譜比較,如圖5所示.R1和R2峰分別對應于最高占據態軌道(HOMOs)中HOMO?1軌道和HOMO?2軌道的共振增強.而HOMO?3的共振增強信號與俄歇信號交疊,無法在譜圖中分辨.285.4 eV對應于苝環碳1s電子到非占據態的躍遷.躍遷軌道與苝環πp軌道貢獻的HOMO?1軌道有空間交疊.因此,HOMO?1軌道在此能量下共振增強.287.6 eV對應于羰基碳1s電子到非占據態的躍遷.這說明HOMO?2軌道有πco軌道的貢獻.這與文獻報道結果一致:HOMO?2軌道來自于πp軌道和酸酐官能團的πco軌道的雜化[25,26].
在共振俄歇退激發過程中(見圖2(f)),假設退激發電子和出射電子來自同一個分子軌道;不考慮LUMO上受激電子的屏蔽作用而引起的分子軌道變化,我們可以利用以下方程估算共振俄歇電子的結合能(EB):EC1s?EB=EB+EK+Φ.其中Φ=4.35 eV為電子能量分析器的功函數;EC1s是碳1s的結合能(見圖3);EK是出射電子的動能.由于已忽略了屏蔽效應的影響,因此利用此方程僅能估算出共振俄歇信號所對應的分子軌道結合能最小值.因此,當分子苝環碳1s電子(EC1s=284.8 eV)激發到LUMOs軌道時,俄歇共振峰(A1)的電子動能值為EK=272.2 eV.代入方程后得到EB>4.1 eV;當羰基碳1s電子(EC1s=288.4 eV)激發到LUMOs軌道時,俄歇共振峰(A2)的電子動能值為EK=273.3 eV,代入方程后得到EB>5.3 eV.將我們的計算結果與價帶譜(60.0 eV)對比發現,共振俄歇退激發過程出射的電子主要來自于PTCDA分子的高結合能分子軌道能級(HOMO?2/HOMO?3).這說明高結合能分子軌道參與的共振俄歇退激發的時間與芯能級空穴的壽命相近,約為6 fs.值得注意的是,在利用共振光電信號的衰減估算界面處的電子從LUMOs軌道到電極導帶的轉移時間時,需排除共振俄歇信號對共振光電信號的貢獻[10,22].

圖5 (網刊彩色)PTCDA薄膜在60.0,285.4和287.6 eV測量的價帶譜,以Au薄膜的費米能級為結合能零點,同時扣除二次諧波激發的碳1s信號Fig.5.(color online)Valence band spectra collected at 60.0,285.4 and 287.6 eV.The binding energy is referred to the Fermi level of clean Au film.The C 1s signals excited by second-order harmonic X-ray are removed.
4 結 論
用OMBD方法制備了PTCDA薄膜,利用基于同步輻射的NEXAFS譜和RPES譜學技術對薄膜電子結構進行了研究,明確了PTCDA分子殼層電子的激發和退激發過程.在RPES譜中,觀察到三類峰結構:二次諧波激發的碳1s信號、共振光電退激發和共振俄歇退激發過程導致的峰結構.通過分析發現,由于PTCDA分子軌道空間分布的差異,分子軌道共振增強效應具有光子能量依賴性;同時,高結合能分子軌道(>4.1 eV)參與共振俄歇退激發過程.實驗結果表明,有機分子薄膜的RPES譜包含碳1s信號、共振光電和共振俄歇信號.對它們的正確歸屬是利用芯能級空穴時鐘譜研究異質界面超快電子轉移動力學的基礎.
[1]Tang M L,Bao Z N 2011Chem.Mater.23 446
[2]Mei J G,Diao Y,Appleton A L,Fang L,Bao Z N 2013J.Am.Chem.Soc.135 6724
[3]Torsi L,Magliulo M,Manoli K,Palazzo G 2013Chem.Soc.Rev.42 8612
[4]Reineke S,Thomschke M,Lüssem B,Leo K 2013Rev.Mod.Phys.85 1245
[5]Hains A W,Liang Z Q,Woodhouse M A,Gregg B A 2010Chem.Rev.110 6689
[6]Zhao J B,Li Y K,Yang G F,Jiang K,Lin H R,Ade H,Ma W,Yan H 2016Nat.Energy1 15027
[7]Ostroverkhova O 2016Chem.Rev.116 13279
[8]Hu Z H,Zhong Z M,Chen Y W,Sun C,Huang F,Peng J B,Wang J,Cao Y 2016Adv.Funct.Mater.26 129
[9]Pan X,Ju H X,Feng X F,Fan Q T,Wang C H,Yang Y W,Zhu J F 2015Acta Phys.Sin.64 077304(in Chinese)[潘宵,鞠煥鑫,馮雪飛,范其瑭,王嘉興,楊耀文,朱俊發2015物理學報64 077304]
[10]Cao L,Wang Y Z,Zhong J Q,Han Y Y,Zhang W H,Yu X J,Xu F Q,Qi D C,Wee A T S 2014J.Phys.Chem.C118 4160
[11]Brühwiler P A,Karis O,M?rtensson N 2002Rev.Mod.Phys.74 703
[12]Zharnikov M 2015J.Electron.Spectrosc.Relat.Phenom.200 160
[13]Cao L,Gao X Y,Wee A T S,Qi D C 2014Adv.Mater.26 7880
[14]Forrest S R 2003J.Phys.Condens.Matter15 S2599
[15]Tautz F S 2007Prog.Surf.Sci.82 47
[16]Guo Y L,Yu G,Liu Y Q 2010Adv.Mater.22 4427
[17]Ou G P,Song Z,Wu Y Y,Chen X Q,Zhang F J 2006Chin.Phys.B15 1296
[18]Cao L,Zhang W H,Chen T X,Han Y Y,Xu G Q,Zhu J F,Yan W S,Xu Y,Wang F 2010Acta Phys.Sin.59 1681(in Chinese)[曹亮,張文華,陳鐵鋅,韓玉巖,徐法強,朱俊發,閆文盛,許楊,王峰2010物理學報59 1681]
[19]Han Y Y,Cao L,Xu F Q,Chen T X,Zheng Z Y,Wan L,Liu L Y 2012Acta Phys.Sin.61 078103(in Chinese)[韓玉巖,曹亮,徐法強,陳鐵鋅,鄭志遠,萬力,劉凌云 2012物理學報61 078103]
[20]Coville M,Thomas T D 1991Phys.Rev.A43 6053
[21]Cao L,Wang Y Z,Zhong J Q,Han Y Y,Zhang W H,Yu X J,Xu F Q,Qi D C,Wee A T S 2011J.Phys.Chem.C115 24880
[22]Cao L,Wang Y Z,Chen T X,Zhang W H,Yu X J,Ibrahim K,Wang J O,Qian H J,Xu F Q,Qi D C 2011J.Chem.Phys.135 174701
[23]Taborski J,V?terlein P,Dietz H,Zimmermann U,Umbach E 1995J.Electron.Spectrosc.Relat.Phenom.75 129
[24]Kikuma J,Tonner B P 1996J.Electron.Spectrosc.Relat.Phenom.82 41
[25]Kera S,Setoyama H,Onoue M,Okudaira K K,Harada Y,Ueno N 2001Phys.Rev.B63 115204
[26]Zahn D R T,Gavrila G N,Gorgoi M 2006Chem.Phys.325 99
PACS:41.60.Ap,79.60.—i,61.05.cj,81.05.FbDOI:10.7498/aps.66.224101
?Corresponding author.E-mail:lcao@hm fl.ac.cn
?Corresponding author.E-mail:guo_yuxian@163.com
Electronic structure of a 3,4,9,10-perylene-tetracarboxylic-dianhydride thin film revealed by synchrotron-based resonant photoemission spectroscopy?
Li Zhi-Hao1)2)Cao Liang2)?Guo Yu-Xian1)?
1)(School of Materials Science and Chemical Engineering,Anhui Jianzhu University,Hefei 230601,China)2)(Anhui Province Key Laboratory of Condensed Matter Physics at Extreme Conditions,High Magnetic Field Laboratory,Chinese Academy of Sciences,Hefei 230031,China)
31 May 2017;revised manuscript
23 August 2017)
The electronic structure of a 3,4,9,10-perylene-tetracarboxylic-dianhydride(PTCDA)thin film is investigatedin situusing synchrotron-based near edge X-ray absorption fine structure(NEXAFS)spectroscopy and resonant photoemission spectroscopy(RPES).The NEXAFS spectroscopy can monitor the electronic transitions from core level to unoccupied states.The CK-edge NEXAFS spectrum of the PTCDA thin film shows four distinct absorption peaks below 290 eV,which are attributed to the transitions from 1s core level of C-atoms in di ff erent chemical environments(perylene core C-atomsvsanhydride C-atoms)into lowest unoccupied molecular orbitals(LUMOs)with π*symmetry.The RPES spectra are collected in the valence band region by sweeping photon energy across the C 1s→π*absorption edge.Three typical features of the C 1s signals excited by second-order harmonic X-ray,resonant photoemission and resonant Auger features are observed in RPES spectra,and are identi fied,relying on the development of kinetic energy of the emitted photoelectrons upon the change of incident photons energy.It is found that the C 1s signals excited by second-order harmonic X-ray are present at high kinetic energy side of spectrum.The kinetic energy of this feature shows photon energy dependence,that is,this feature shifts to higher kinetic energy by photon energy increasing twice.Resonant Auger peaks in RPES spectra are located on the low kinetic energy side with constant kinetic energy regardless the change of photon energy.The resonant Auger may originate from deeper molecular orbitals with binding energy large than 4.1 eV,suggesting that the resonant Auger decay process involved in deeper molecular orbitals occurs on a time scale comparable to C 1s core hole lifetime of 6 femtoseconds.Resonant enhancement of highest occupied molecular orbitals(HOMOs)derived valence band features or HOMO?1 and HOMO?2 derived resonant photoemission features in our case are lying between the C 1s signals and the resonant Auger signals.The Kinetic energy increases as the photon energy sweeps across the absorption edge,whereas their binding energy remains constant.In addition,the enhancements of two resonances show photon energy dependence that enhancement of HOMO?1 related resonance dominates over HOMO?2 related resonance at energies corresponding to perylene core C 1s to LUMOs transitions,whereas HOMO?2 related resonance becomes dominant at transitions from anhydride C 1s to LUMOs.This behavior
*Project supported by the National Natural Science Foundation of China(Grant Nos.11574317,21503233),Anhui Provincial Natural Science Foundation,China(Grant No.1608085MA07),and the Natural Science Foundation from the Education Bureau of Anhui Province,China(Grant No.KJ2016A143). can be related to the wavefunction character and symmetry of the frontier molecular orbitals.Clarifying each resonant feature in RPES spectra and their origin will pave the way for accurately determining the ultrafast charge transfer time at organic/electrode interfaces using synchrotron-based core hole clock technique implementation of RPES.
organic semiconductor,near edge X-ray absorption fine structure,Auger resonance,synchrotron-based photoelectron spectroscopy
10.7498/aps.66.224101
?國家自然科學基金(批準號:11574317,21503233)、安徽省自然科學基金(批準號:1608085MA07)和安徽高校自然科學研究項目(批準號:KJ2016A143)資助的課題.
?通信作者.E-mail:lcao@hm fl.ac.cn
?通信作者.E-mail:guo_yuxian@163.com
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
