縮醛化反應合成聚甲氧基二甲基醚研究進展
2017-12-22 05:36:34金福祥宋河遠康美榮夏春谷陳靜
化工學報 2017年12期
關鍵詞:催化劑
金福祥,宋河遠,2,康美榮,夏春谷,陳靜
(1中國科學院蘭州化學物理研究所,羰基合成與選擇氧化國家重點實驗室,甘肅 蘭州 730000;2中國科學院大學,北京 100049)
縮醛化反應合成聚甲氧基二甲基醚研究進展
金福祥1,宋河遠1,2,康美榮1,夏春谷1,陳靜1
(1中國科學院蘭州化學物理研究所,羰基合成與選擇氧化國家重點實驗室,甘肅 蘭州 730000;2中國科學院大學,北京 100049)
聚甲氧基二甲基醚具有很高的十六烷值和含氧量,低冷凝點和冷濾點,能顯著改善柴油的燃燒特性,有效地提高熱效率,大幅度減少NOx和碳煙的排放,被認為是一種優良的環保型燃油組分。綜述了近年來縮醛化反應合成聚甲氧基二甲基醚催化劑體系、反應機理、反應動力學研究進展,已報道的催化劑體系包括液體酸催化劑、固體酸催化劑及離子液體催化劑體系,涉及的反應原料包括可提供亞甲氧基的化合物,如甲醛水溶液、多聚甲醛、三聚甲醛,以及提供封端甲基的化合物,如甲醇、甲縮醛、二甲醚等甲醇下游產品。另外,綜述了國內外縮醛化反應合成聚甲氧基二甲基醚反應動力學研究進展,及所建立的反應動力學模型和得到的動力學參數。縮醛化反應及動力學研究正朝著更有利于工業化生產的方向發展,將對工業化生產具有一定的參考意義。
聚甲氧基二甲基醚;催化劑;液體酸;固體酸;分子篩;離子液體;動力學
引 言
聚甲氧基二甲基醚 (polyoxymethylene dimethyl ether,CH3O(CH2O)nCH3,DMMn或 PODEn)是一種多醚類化合物,當 DMMn分子中亞甲氧基數n≤3時,在常溫下為無色透明、易揮發的液體,具有氯仿氣味,能溶于3倍于其體積的水,易與醇類和醚類互溶。當n≥5 時,在常溫下呈白色的固體。DMMn具有很高的十六烷值(CN值:DMM263,DMM378,DMM490,DMM5100)和含氧量(含氧量為42%~49%),閃點為 64~84℃,符合柴油安全標準,沸點為156~268℃,正好處于柴油范圍內[1]。同時,由于分子結構中存在活潑的亞甲氧基 (—CH2O—),在燃燒過程中易生成過氧化氫中間體 HO2?,HO2?分解生成羥基自由基 HO?,HO?進行連續氧化反應進而有效地降低了碳煙、CO和烴類化合物的排放。因此,DMMn被認為是一種優良的環保型柴油調和組分。據報道[2-5],柴油中添加 15%(體積分數)的 DMM3—6,在不影響柴油煙點、滯燃期等性能的情況下,柴油的潤滑性明顯提高,同時尾氣中NOx、顆粒物和碳氫化合物的排放達到了歐Ⅴ的標準。DMMn具有優異的溶解性和極強的滲透能力,能與許多有機溶劑互溶,低毒,可替代苯、甲苯、二甲苯等工業溶劑,也可廣泛應用于工業溶劑、顏料分散劑。
DMMn的傳統合成方法通常在酸性條件下,采用可提供亞甲氧基的化合物(如甲醛水溶液、多聚甲醛、三聚甲醛等)和封端甲基的化合物(如甲醇、甲縮醛、二甲醚等)縮醛化反應制備,反應式見圖1[6]。已報道的催化劑體系主要有液體酸、固體酸、離子液體等。其他的合成路線如合成氣直接合成法[7]、甲醇選擇氧化法[8-10]、二甲醚催化氧化法[11]等。近年來,隨著煤化工的發展,以煤為原料合成甲醇的技術已經成熟,國內甲醇的產能呈高速增長的局面。2016年,我國甲醇的年產能已達7500萬噸,由于下游產品開發不夠,導致甲醇產能過剩,因此推動開發甲醇下游產品并向高附加值發展對于煤化工的健康發展具有深遠意義。DMMn產業鏈流程如圖 2所示,甲醇與甲醛或聚甲醛反應合成DMMn,充分保留了原料中的氧原子,使甲醇的利用率達到 80% 以上。
本文綜述了近年來縮醛化反應合成聚甲氧基二甲基醚催化劑體系、反應機理及動力學研究進展,已報道的催化劑包括液體酸、固體酸、離子液體等,并對聚甲氧基二甲基醚的發展及應用前景進行了展望。
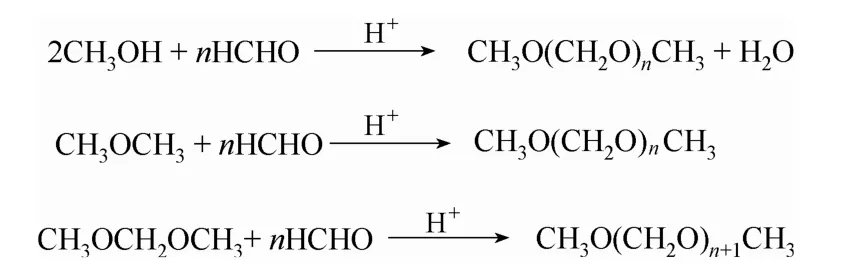
圖1 縮醛化反應制備DMMn反應式Fig.1 Production of DMMn by acetalation of formaldehyde
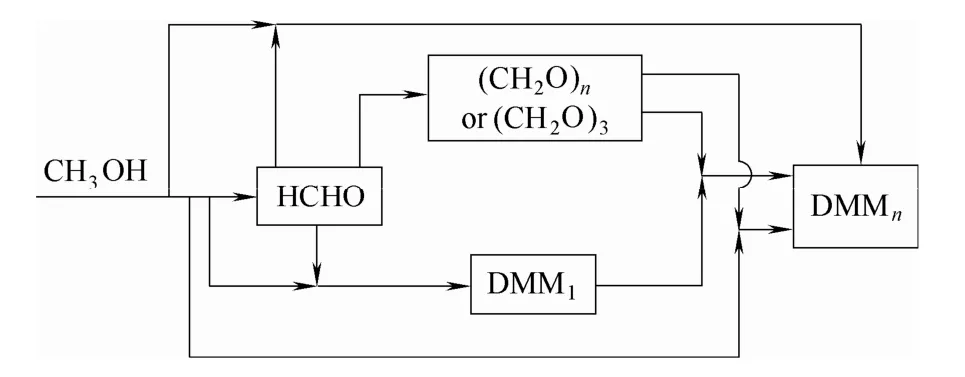
圖2 DMMn產業鏈流程Fig.2 Block flow diagram of DMMn process chain
1 催化劑研究進展
1.1 液體酸催化劑體系
縮醛化反應合成聚甲氧基二甲基醚的研究可以追溯到20世紀20年代。1925 年,研究者通過硫酸或鹽酸催化甲醇與多聚甲醛反應,在較苛刻的條件下(150~180℃,12~15 h)反應合成了低分子量的 DMMn,反應過程中有副產物 CO2生成[12]。Willian等[13]以無機酸(如H2SO4)為催化劑,甲縮醛(DMM1)或甲醇與低聚甲醛在較溫和的條件下[催化劑量為 0.1%(質量分數),DMM1:HCHO=4:1(摩爾比),100℃,1h]反應,主要得到了低聚合度的DMM2—4。Moulton等[2]報道了甲酸催化甲縮醛與多聚甲醛的縮醛化反應,在甲酸用量為0.1%(質量分數),DMM1:HCHO=1:5(摩爾比)的條件下反應得到了DMM1—10的混合物。
2001年以來,Patrini等[6]申請了一系列液體酸催化縮醛化反應合成DMMn的反應及工藝專利,以CF3SO3H 為催化劑,在 CF3SO3H:HCHO:DMM1=1:5.5:7.3 (摩爾比),115℃的條件下反應 40 min,DMM2—5的收率達到了 50%以上。隨后, Stroefer等[14]以H2SO4為催化劑,研究了三聚甲醛(TOX)和二甲醚(DME)的縮醛化反應,考察了磺酸(CF3SO3H、p-toluenesulfonic acid)、雜多酸等催化劑的催化性能,申請了液體酸催化劑體系的系列工藝專利[15-18]。2013年,Zhao等[19]以 H2SO4、磺酸化改性的酚醛樹脂及酸性樹脂D008為催化劑,研究了甲縮醛和多聚甲醛縮醛化反應機理(圖3)。研究發現,反應產物分布遵循Schulz-Flory分布規則(圖4),并提出了鏈增長反應機理,得到了鏈增長反應方程[式(1)]。


圖3 多聚甲醛的分解及鏈增長合成DMMn反應機理Fig.3 Decomposition of paraformaldehyde and chain propagation for synthesis of DMMn

圖4 不同溫度下甲縮醛和多聚甲醛反應合成DMMn的Schulz-Flory分布Fig.4 Schulz-Flory distribution of DMMn synthesized from reaction between dimethoxymethane and paraformaldehyde at different temperatures
雖然研究人員對液體酸催化反應工藝過程進行了不斷的改進和優化,然而液體酸催化劑存在設備腐蝕嚴重、催化劑分離困難、不能循環使用、后處理能耗大等缺陷。合成單元關鍵技術未能取得突破,引發了人們對新的綠色催化體系的探討。
1.2 固體酸催化劑體系
研究者們采用固體酸催化劑取代傳統的液體酸,因此固體酸催化劑成為了研究的熱點之一。BP公司申請了一系列固體酸催化劑體系(如離子交換樹脂、分子篩等)催化縮醛化合成DMMn的專利,并對反應及分離工藝進行了研究[20-26]。隨后國內外研究人員在該領域開展了大量的研究工作,已報道的固體酸催化劑體系主要有離子交換樹脂、分子篩、金屬氧化物、石墨烯等。
1.2.1 樹脂類催化劑 早在1987年,Arvidson等[27]就報道了酸性陽離子交換樹脂 Amberlite IR120催化甲醛與甲縮醛反應合成DMMn的研究,并考察了鹵化鋰助劑對反應的影響,對助劑的作用機制進行了推導(圖 5)。在 DMM1:HCHO=2:1(摩爾比)、100℃的條件下反應24 h,DMM2—4在反應液中的含量達到了 33.0%。當以多聚甲醛作為甲醛源時,反應活性優于三聚甲醛,他們認為這主要是由于三聚甲醛的環狀結構難以解聚造成的。
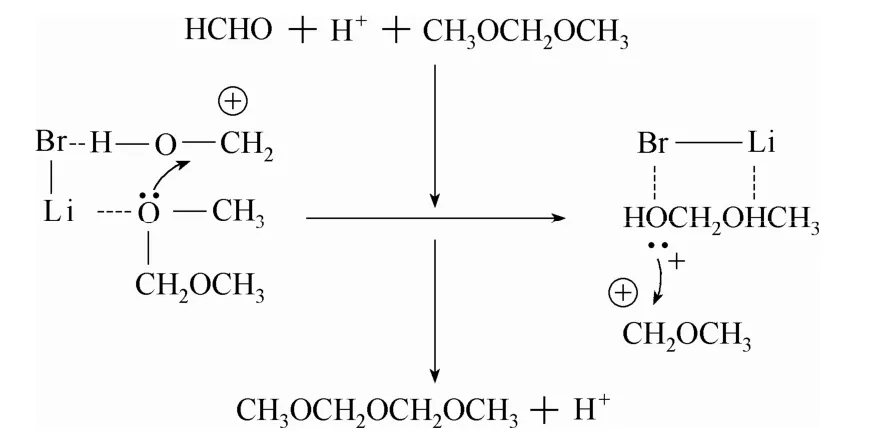
圖5 鹵化鋰助催化反應的機理Fig.5 Mechanism of catalytic reaction with help of lithium halides
2008年,Stroefer 等[14]以 Amberlite IR120 離子交換樹脂為催化劑,考察了三聚甲醛和二甲醚的縮醛化反應,一定工藝條件下主要得到DMM2—4。隨后 Clarisse等[28]、Burger等[29]相繼報道了 Amberlyst A15、Amberlyst 36陽離子交換樹脂催化甲縮醛和三聚甲醛的縮醛化反應。在 DMM1:TOX=0.25:1(摩爾比)、50℃反應條件下,DMM2的收率最高達到了50%以上,DMM3—4的收率僅為30%。此外,他們對三聚甲醛與甲縮醛反應機理進行了研究,如圖 6所示。認為反應可能有兩種模式。模式 1:三聚甲醛首先在酸性條件下分解為甲醛,甲醛分子作為DMMn的聚合單體與甲縮醛在 H+作用下反應生成DMM2;同樣,DMMn與甲醛單體反應生成DMMn+1。模式2:三聚甲醛環只斷裂一個C—O鍵,然后與甲縮醛反應生成了DMM4,該反應模式有利于長鏈多醚 (DMMn,n=4,7,10,…) 的合成。

圖6 合成 DMMn 反應機理Fig.6 Reaction scheme of DMMn formation
劉現立等[30]考察了大孔強酸性陽離子交換樹脂催化性能,在催化劑用量 5%(質量分數)、DMM1:HCHO=1(摩爾比)、90℃、1.0 MPa的條件下反應6 h,HSD001表現出了最好的催化性能,甲縮醛的轉化率達到56%。類似地,陳婷等[31]報道了大孔強酸性陽離子交換樹脂 CT175催化劑,在CT175用量 7.5%(質量分數)、DMM1:TOX=3:1(摩爾比)、90℃、1.5 MPa的條件下反應0.5 h,三聚甲醛的轉化率為 89.0%,DMM3—8的選擇性為 64.2%。催化劑重復使用20次后,催化活性無明顯變化。Zheng等[32]考察了不同類型大孔陽離子交換樹脂的催化性能,結果如表1所示,具有較大比表面積、孔容和孔徑的NKC-9表現出了最好的催化活性,甲醛的轉化率為84.7%。他們認為NKC-9由于具有較高的交換性能、適當的孔結構,增加了催化劑與原料的接觸,有利于催化作用的發生。此外,對反應機理進行了推導(圖7),首先多聚甲醛分解為甲醛,甲醛經催化劑酸性位質子化生成羰基碳正離子,然后與DMMn發生親電加成反應生成DMMn+1。

表1 不同催化劑合成DMMn的產物分布Table 1 Product distribution (SDMMn) over different catalysts
隆寬燕等[33]報道了大孔強酸性樹脂 HD-8催化劑體系,在催化劑用量 2.2%(質量分數)、CH3OH:TOX=1:2(摩爾比)、2.0 MPa、110℃條件下反應3 h,DMM3—8的選擇性達到了77.54%,收率為 57.42%。2013年,施敏浩等[34]采用 AlCl3[負載量為 2.5%(質量分數)]對大孔強酸性陽離子交換樹脂進行了改性,以單因素實驗和正交實驗相結合的方式,系統地研究了甲醛與甲醇縮醛化反應。得到甲醇的轉化率為 69.72%,DMM3—8的選擇性為62.08%。此外,劉顯科等[35]報道了對甲苯磺酸改性的大孔陽離子樹脂的催化反應,在 DMM1:HCHO=1:4(摩爾比)、70℃、1.5 MPa、重時空速為 3 h-1的條件下反應0.5 h,DMM1的轉化率為60.54%,DMM3—5的選擇性和收率分別為 33.92%和 20.54%。時米東等[36]采用氯化鋅對強酸性大孔樹脂進行了改性,在催化劑用量 10%(質量分數)、CH3OH:HCHO=0.5:1(摩爾比)、105℃的條件下反應3 h,反應液中DMM1—8的含量可達到 40%以上,DMM2—8選擇性達到60%以上。

圖7 固體酸催化合成DMMn的反應機理Fig.7 Scheme of heterogeneous acid catalytic mechanism for DMMn synthesis
1.2.2 分子篩類催化劑 2010年,馮偉樑等[37]報道了β沸石、Y型分子篩等催化劑體系,在催化劑HY用量為 1.1%(質量分數)、CH3OH:TOX=2.2:1(摩爾比)、150℃、1.0 MPa的條件下反應4 h,三聚甲醛的轉化率為91.8%,DMM3—10選擇性為50.2%。李豐等[38]報道了基于一定硅鋁比的不同類型催化劑(β沸石、X型沸石、Y型沸石、ZSM-5分子篩、MCM-22分子篩等)催化甲縮醛或甲醇與多聚甲醛的縮醛化反應。在催化劑用量為1.1%(質量分數)、130℃、0.5 MPa的條件下反應4 h,甲醛的轉化率達到了 94.2%,DMM3—10選擇性為50.8%。該研究團隊還報道了固體超強酸負載的分子篩催化劑,在催化劑用量0.3%(質量分數)、CH3OH:TOX=2.2:1(摩爾比)、130℃、0.7 MPa 的條件下反應4 h,三聚甲醛的轉化率為92.4%,DMM3—10選擇性為55.2%[39]。
Zhao等[40]考察了 HY、HZSM-5、Hβ和HMCM-22等分子篩結構及酸強度對縮醛化反應性能的影響,結果見表 2。研究發現,表面弱酸位有利于短鏈產物DMM1的生成,如HY;但是酸性太強,則不利于長鏈產物的穩定存在,從而影響DMMn的收率,如 HZSM-5。而具有中等強度酸性的HMCM-22則能促進 DMM3—8的生成,同時HMCM-22分子篩具有較高的比表面積和孔體積,并具有超籠結構,這種結構有利于生成分子尺寸較大的DMM3—8。以HY為催化劑時,反應產物主要為 DMM1—3;以 HZSM-5 和 Hβ 為催化劑時,DMM3—8的收率分別為 6.40%和 13.78%;當以HMCM-22 為催化劑時,長鏈的聚合物收率明顯增加,DMM3—8的收率可以達到29.39%。隨后該研究團隊還考察了HMCM-22 分子篩中硅鋁比(Si/Al)對甲縮醛與三聚甲醛縮醛化反應的影響,當Si/Al=200,催化劑表現出了較高的活性。另外,曹健等[41]也研究了分子篩孔道結構、酸位等對反應的影響。研究發現HMCM-22分子篩具有六方片狀晶粒結構,晶粒粒徑約為 0.8 μm×1 μm×0.05 μm,其內部Br?nsted酸位比例最高,所以表現出了最好的催化活性。在催化劑用量為 5%(質量分數)、CH3OH:TOX=2:1(摩爾比)、120℃的條件下反應 10 h,三聚甲醛轉化率為39.8%,DMM2—8的選擇性最高達到了65.1%。以MOR分子篩為催化劑時,在同等條件下反應,DMM2—8的選擇性僅為23.2%(表3)。他們認為,催化劑中強的 Br?nsted酸位是催化縮醛化反應的主要因素,分子篩中強酸位可以促進DMMn中長鏈分子的生成。但是分子篩孔道增加并不利于DMMn的生成,DMMn生成過程與孔道結構沒有直接關系。
2014年,張向京等[42]通過在HMCM-22分子篩上負載磷鎢酸(PW)來增強催化劑的酸性,從而增強其催化反應活性。考察了磷鎢酸的負載量、焙燒溫度和焙燒時間等對催化性能的影響。當PW負載量為30%(質量分數),300℃焙燒4 h時,表現出了最高的催化活性。在 CH3OH:TOX=1.2:1(摩爾比)、催化劑用量為3%(質量分數)、150℃的條件下反應6 h。三聚甲醛轉化率為87.1%,DMM2—8的選擇性為54.8%。
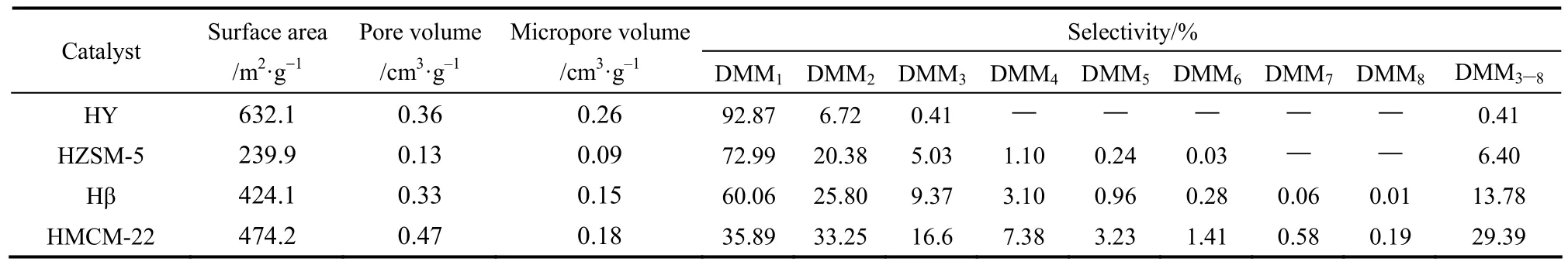
表2 不同分子篩催化甲醇與三聚甲醛縮醛化反應性能Table 2 Catalytic performances of different molecular sieves on acetalation of methanol and trioxymethylene

表3 MOR、Al-YNU-1和HMCM-22分子篩催化甲醇與三聚甲醛的縮醛化反應Table 3 MOR,Al-YNU-1 and HMCM-22 for synthesis of DMMn with methanol and trioxymethylene
2015年,Wu等[43]考察了HZSM-5分子篩Si/Al比對縮醛化反應的影響,結果見表4。當Si /Al=580時表現出了最好的催化活性,三聚甲醛轉化率為85.3%,DMM2—8的選擇性為88.5%。同時催化劑表現出了較好的穩定性,循環使用 15次,DMM2—8的收率未見明顯降低。他們認為,HZSM-5催化劑上適當 Si/Al比可以提供充足的酸位點,有效地促使三聚甲醛分解為甲醛,同時也能夠促進甲醛與甲縮醛反應,從而使短鏈 DMMn更有利于形成長鏈DMMn(圖8)。而隨著 Si/Al比的升高強酸位點會逐步減少,這也會抑制甲醛轉化為副產物(甲酸甲酯)的反應。此外,高曉晨等[44]采用P2O5對HZSM-5分子篩進行了改性,當Si /Al=50,粒徑尺寸為5 μm,P2O5含量≤6%(質量分數)時,表現出較高的催化活性和DMMn選擇性。在催化劑用量為1%(質量分數)、CH3OH:TOX=5.5:1(摩爾比)、130℃的條件下反應,三聚甲醛轉化率可達到 95.2%,DMM2—5的選擇性為 62.9%。
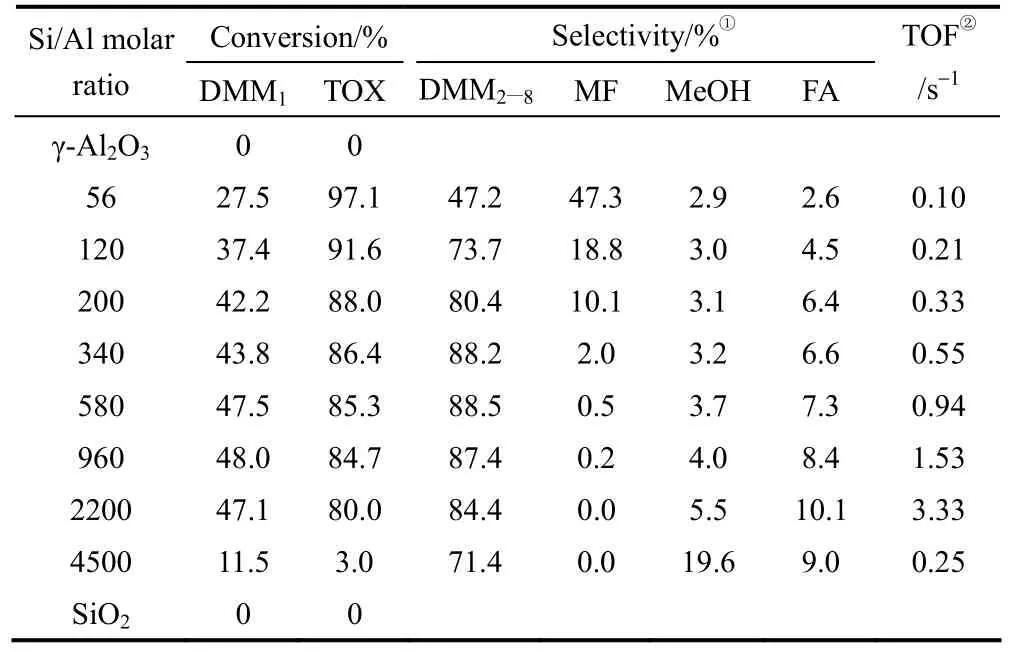
表4 HZSM-5分子篩不同硅鋁比對縮醛化反應的影響Table 4 Catalytic performance of HZSM-5 zeolites with various Si/Al ratios in synthesis of DMMn
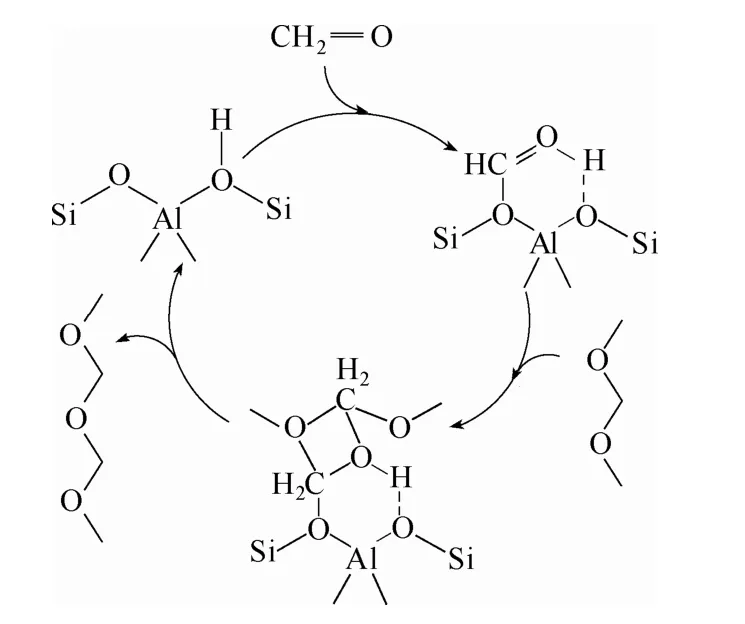
圖8 HZSM-5分子篩催化縮醛化反應合成DMMnFig.8 Proposed reaction pathways for synthesis of DMMn by HZSM-5 zeolite
何欣等[45]制備了一種晶粒直徑為0.2 μm,硅鋁比為30的ZSM-5分子篩,在催化劑用量為0.5%(質量分數)、CH3OH:HCHO:DME=1:10:5.5(摩爾比)、110℃、3.0 MPa的條件下反應3 h,甲醛轉化率為81.5%,DMM3—8的選擇性為 32.4%。2015年,劉志成等[46]報道了金屬離子(Sn、Mn、Cu、Ti等)改性的氫型強酸性分子篩催化反應性能,在催化劑用量 4.0%(質量分數)、DMM1:CH2O=0.8:1(摩爾比)、115℃、0.6 MPa的條件下反應6 h,DMM3—10的選擇性達到了63%。
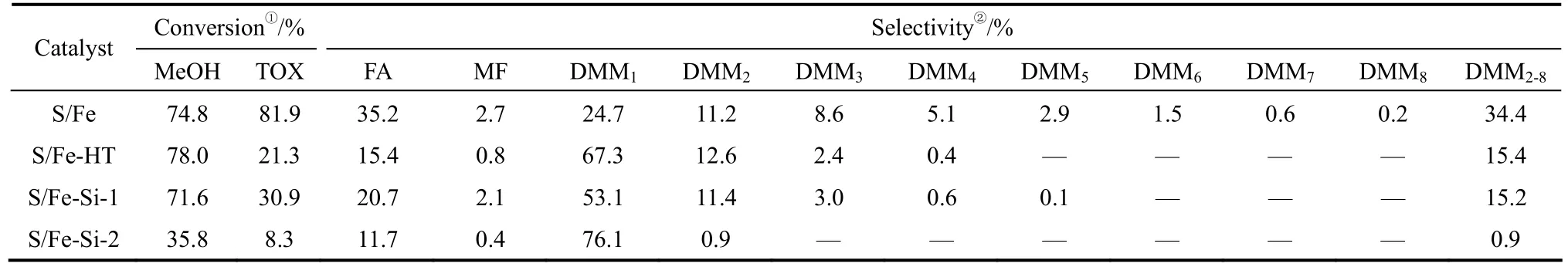
表5 合成DMMn的產物分布及催化活性Table 5 Catalytic activity and DMMn products distributions by acetalation reaction
1.2.3 金屬氧化物類催化劑 2010年,李豐等[47]將H2SO4、HCl負載在金屬氧化物(ZrO2、TiO2等)上制得固體超強酸催化劑,在催化劑用量為 1.1%(質量分數)、CH3OH:TOX=2.2:1(摩爾比)、130℃、0.7 MPa的條件下反應 4 h,三聚甲醛的轉化率為95.0%,DMM3—10選擇性為 54.4%。同年,洪正鵬等[48]報道了經TiO2改性的γ-Al2O3-TiO2催化甲醇和甲醛的縮醛化反應。在 CH2O:CH3OH=10:1(摩爾比)、80℃、4.5 MPa、液體體積時速(LHSV)為0.5 h-1的條件下反應,甲醇轉化率為94.0%,反應液中DMM3—8的含量達到了69.3%。2014年,Zhang等[49]制備了一系列Al2O3/ZrO2復合氧化物催化劑,當催化劑中ZrO2的含量達到4%(Al2O3-4Zr)時,表現出了最高的催化活性,甲醇的轉化率為50%,DMM3—8選擇性為 20%。趙峰等[50]報道了固體酸催化劑體系,在催化劑用量1.5%(質量分數)、CH3OH:TOX=1.2:1(摩爾比)、130℃、0.1 MPa條件下反應2 h,DMM3—8的收率為22%。他們認為由于的部分流失對催化劑的弱酸位影響不大,從而使具有良好的循環活性。2015年,Li等[51]制備了不同酸強度和 Br?nsted酸位的固體酸,考察了催化劑中不同的表面酸性對反應性能的影響(表5)。具有較高酸密度及Br?nsted酸位的S/Fe表現出了最高的催化活性,三聚甲醛的轉化率為 81.9%,DMM2—8的選擇性為 34.4%。認為催化劑活性主要取決于表面酸性的強弱,反應選擇性取決于催化劑酸性強弱和催化劑上的 Br?nsted酸位置。隨后,Li等[52]報道了催化甲縮醛和三聚甲醛的縮醛化反應,三聚甲醛轉化率達到了 89.5%,DMM2—8選擇性為54.8%。催化活性優于單獨使用H2SO4或TiO2。
1.2.4 其他催化劑 2012年,沈儉一等[53]報道了酸性碳材料(強酸量≥1.0 nmol·g-1)催化縮醛化反應合成聚甲氧基二甲醚的方法,以甲縮醛和三聚甲醛為原料,在催化劑用量 0.2%(質量分數)、MM1:CH2O=1:1(摩爾比)、120℃條件下反應 2 h,反應液中DMM2—8的含量為44.6%。此外,高曉晨等[54]報道了活性炭催化劑體系(BET比表面積為1000~2500 m2·g-1,孔容為 0.3~0.6 ml·g-1),DMM2—10的選擇性最高可達到77.0%。
2014年,王一萌等[55]報道了具有Pm3n對稱結構的超微孔硅鋁催化材料在甲縮醛和三聚甲醛縮醛化反應中的應用,催化劑孔徑為1.3~2.0 nm,在催化劑用量為 7.5%(質量分數)、DMM1:TOX=3:1(摩爾比)、1.3 MPa、105℃條件下反應2 h,三聚甲醛的轉化率最高可達到92.1%,反應液中DMM3—8的含量為53.6%。隨后,Fu等[56]系統考察了對稱超微孔硅鋁催化劑結構與產物分布的關系,結果發現,改變催化劑孔徑的大小使得 DMMn分子流動受到限制,從而改變了DMMn合成的產物分布情況(圖9)。當以具有Pm3n對稱結構C10-AS-50為催化劑時,其孔徑等于 DMM8分子直徑,使得 DMM8分子擴散受到局部限制,DMM3—8表現出較高的選擇性(表6)。三聚甲醛轉化率達到 92.1%,DMM3—8的含量達到53.5%。
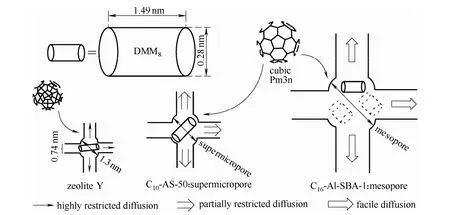
圖9 不同孔道的硅鋁酸鹽催化劑對DMMn(如DMM8)選擇性的影響說明Fig.9 Illustration of DMMn product selectivity (DMM8 as example) in pores with increased diameters of different aluminosilicate catalysts

表6 不同催化劑結構及反應結果Table 6 Characterization and reaction results of different catalysts
2015年,Fang等[57]報道了聚乙烯吡咯烷酮-硅鎢酸(PVP-HSiW)和聚乙烯吡咯烷酮-磷鎢酸(PVP-HPW)催化劑體系催化甲醇和三聚甲醛的縮醛化反應(表7)。當以PVP-HSiW(配比為1/4:3/4)為催化劑時,DMM2—5的選擇性為 52.5%,轉化率為95.7%;當以PVP-HPW(配比為1/4:1)為催化劑時,DMM2—5的選擇性達到了 54.9%,轉化率為95.4%。王建國等[58]報道了氧化石墨烯在縮醛化反應的應用,石墨烯比表面積>65 m2·g-1,C/O摩爾比為0.8~1.5,單層厚度為0.6~0.8 nm,橫向尺寸為200 nm~10 μm。在催化劑用量為1.0%~5.0%(質量分數)、CH3OH:TOX=(0.1~10):1(摩爾比)、75~150℃、常壓條件下反應1~15 h,三聚甲醛的轉化率最高達到了77.9%,DMM2—8的選擇性為68.9%。類似地,Wang等[59]考察了氧化石墨烯處理過程對其催化活性的影響(表8),結果發現,在氧化石墨烯催化劑混合制備過程中,反應時間為10 h時,其催化效果最好。甲醇和三聚甲醛的轉化率分別達到90.3%和92.8%,DMM2—8的選擇性為30.9%。
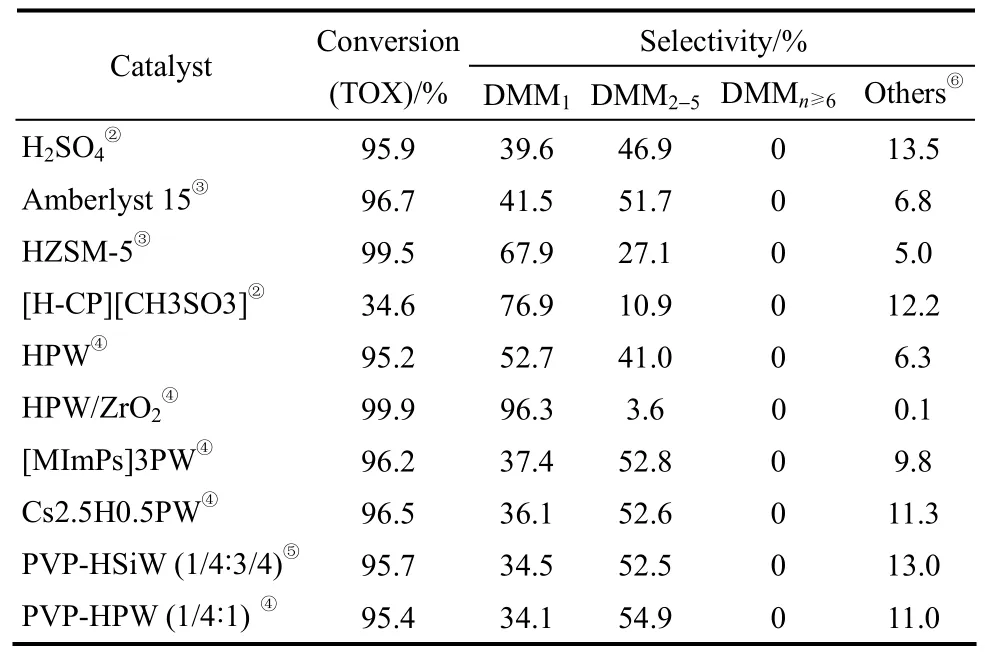
表7 不同酸性催化劑催化反應結果Table 7 Results of using various acidic catalysts①
1.3 離子液體催化劑體系
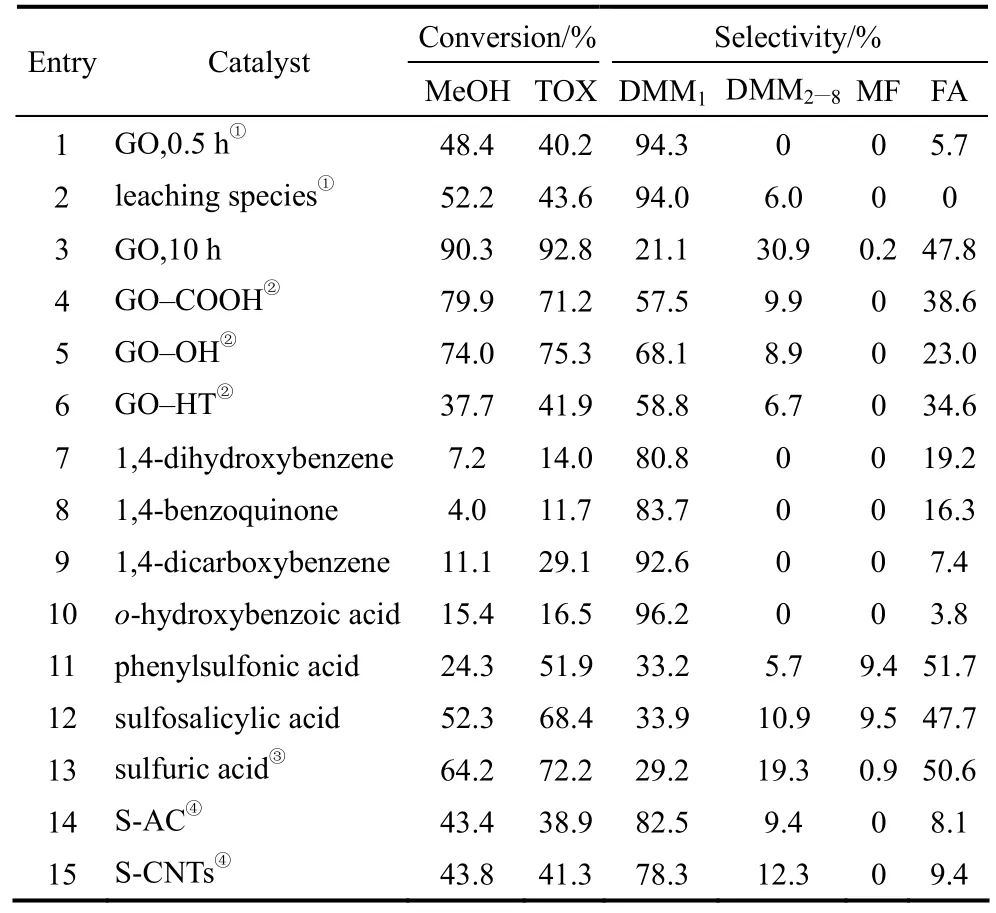
表8 不同催化劑催化合成DMMn的反應結果Table 8 Results for synthesis of DMMn over various catalysts
離子液體是一種由陰陽離子構成的、熔點低于100℃的液態熔鹽,是一類新型的介質和軟功能材料,具有良好的熱穩定性和化學穩定性。近年來,利用離子液體的可設計性開展的應用研究受到人們的普遍關注。自2006年起,本課題組最先開展了離子液體催化縮醛化反應合成DMMn的研究,并申請了系列催化反應及工藝專利[60-70]。離子液體陽離子包括羧酸或磺酸功能化的咪唑陽離子、吡啶陽離子等,陰離子選自對甲基苯磺酸根、三氟甲基磺酸根等(圖 10),研究了離子液體催化劑分離方式與重復使用情況。以甲醇為初始反應原料,開發了甲醇-三聚甲醛、甲醇-多聚甲醛、甲縮醛-三聚甲醛、甲縮醛-多聚甲醛等反應體系,在一定條件下反應,三聚甲醛轉化率≥90%,DMM3—8選擇性>43%。以甲縮醛和三聚甲醛為反應原料,三聚甲醛轉化率 > 90%,DMM3—8選擇性>50%。催化劑多次循環使用催化活性不變。在反應研究基礎上,采用密度泛函理論(DFT)計算的方法對酸性離子液體催化合成DMMn反應微觀過程進行了研究[71]。首先,三聚甲醛在酸性離子液體存在下經兩個基元步驟解聚為甲醛單體,其解聚的勢能面圖如圖11所示。三聚甲醛通過O…H—O氫鍵與SO3H-FIL生成絡合物IM1需要吸收1.78 kcal·mol-1的熱量,經過過渡態TS1-2,生成類半縮醛的鏈狀三聚甲醛IM2,該過程需要克服的能壘為20.22 kcal·mol-1。通過觀察解聚反應的逆反應,發現需要克服的能壘高出2.02 kcal·mol-1,這說明 TOX的解聚過程是一個平衡過程,加熱對反應有利。其次,分別對甲醇或甲縮醛與甲醛在酸性離子液體催化下的反應過程進行了量化計算。DMM1與甲醛的反應遵循“碳正離子機理”,即DMM1被離子液體催化劑質子化,釋放出甲醇的同時生成碳正離子中間體 C1+,甲醛進攻碳正離子實現鏈增長,與甲醇反應得到不同鏈長的 DMMn產物。此外,研究人員將離子液體嫁接到硅膠上,制備了硅膠負載的雙重酸性離子液體催化劑,制備過程如圖12所示。在催化劑用量為4.0%(質量分數)、DMM1:TOX=3:1(摩爾比)、105℃條件下反應 1 h,三聚甲醛轉化率>90%,DMM3—8的選擇性>50%。研究結果表明,催化劑酸密度是影響催化活性的主要因素[72]。
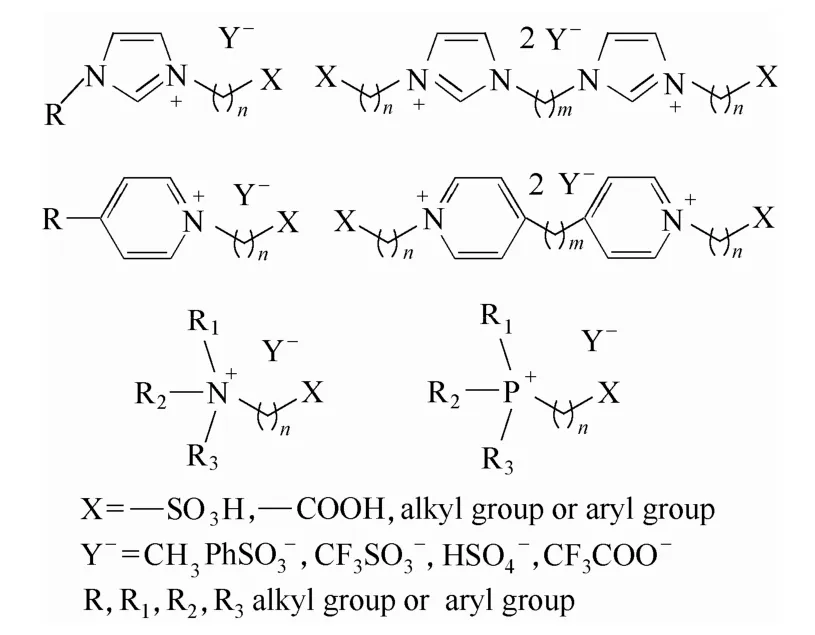
圖10 功能化離子液體結構式Fig.10 Structures of functional ionic liquids
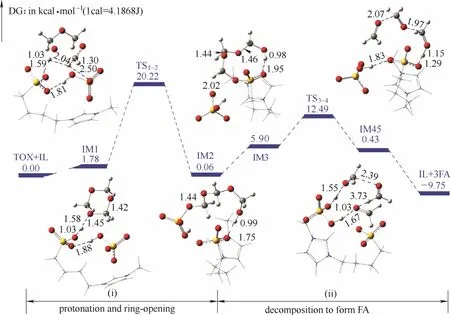
圖11 TOX解聚勢能面圖Fig.11 Energy surface for decomposition of 1,3,5-trioxane (TOX)
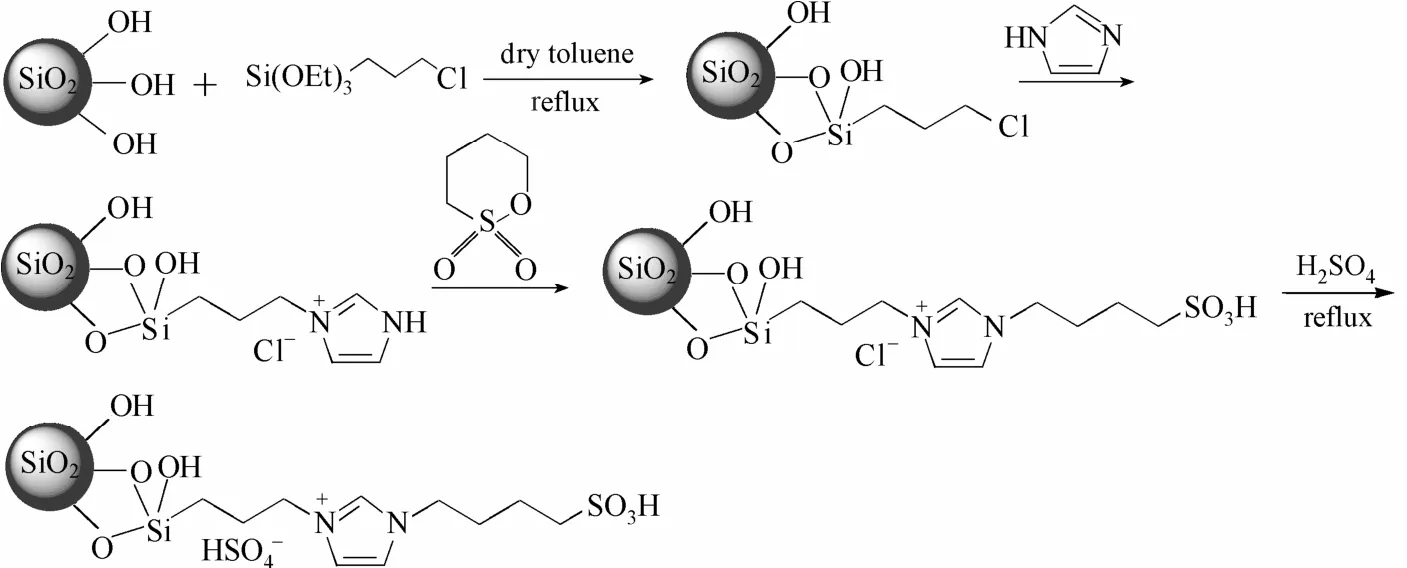
圖12 通過嫁接方法合成硅膠負載離子液體Fig.12 Synthesis of silica-gel-supported ionic liquids (ILs) via grafting method
2013年,趙變紅等[73]考察了不同磺酸功能化離子液體催化縮醛化反應,在催化劑用量為2.1%(質量分數)、TOX:CH3OH=1:2(摩爾比)、120℃、2.0 MPa的條件下反應4 h,三聚甲醛的轉化率達到了90%以上,DMM3—8的選擇性達到了60.95%。類似地,Wu等[74]報道了磺酸功能化離子液體催化甲縮醛和三聚甲醛的縮醛化反應,在 DMM1:TOX:ILs=180:60:1(摩爾比)、170℃條件下反應 10 h,[PyBS]HSO4具有較高催化活性,TOX轉化率為91.2%,DMM3—8選擇性達到了70.9%。他們認為離子液體酸度越高催化活性越好。隨后該研究團隊[75]考察了磺酸功能化離子液體陽離子上烷基取代基對其催化活性的影響,研究發現隨著離子液體N原子上烷基碳鏈的增長,離子液體的疏水性增加,同時DMM3—8的選擇性先增加后下降。其中[C6ImBS]HSO4表現出較高的活性,DMM3—8選擇性最高達到了57.8%。趙強等[76]報道了N-甲基-2-吡咯烷酮硫酸氫鹽[Hnmp]HSO4和N-甲基-2-吡咯烷酮對甲苯磺酸鹽[Hnmp]PTSA 催化劑體系,在[Hnmp]HSO4用量為 2.0%(質量分數)、DMM1:CH2O=0.8:1(摩爾比)、110℃條件下反應時間6 h,甲縮醛的轉化率為52.28%,DMM3—8的選擇性為 49.18%。類似地,李為民等[77]報道了基于N-甲基吡咯烷酮的酸功能化離子液體催化劑體系。在一定條件下反應,[PyN(CH2)3SO3H]HSO4表現出了較高的催化活性(表 9),三聚甲醛的轉化率和DMM3—8的選擇性分別為 97.69%和 32.54%。鄧小丹等[78]報道了復合型離子液體催化三聚甲醛和甲醇的反應,在復合型催化劑用量為5%(質量分數)、TOX:CH3OH=0.5:1(摩爾比)、100℃、2.0 MPa下反應4 h,三聚甲醛轉化率為 96.66%。
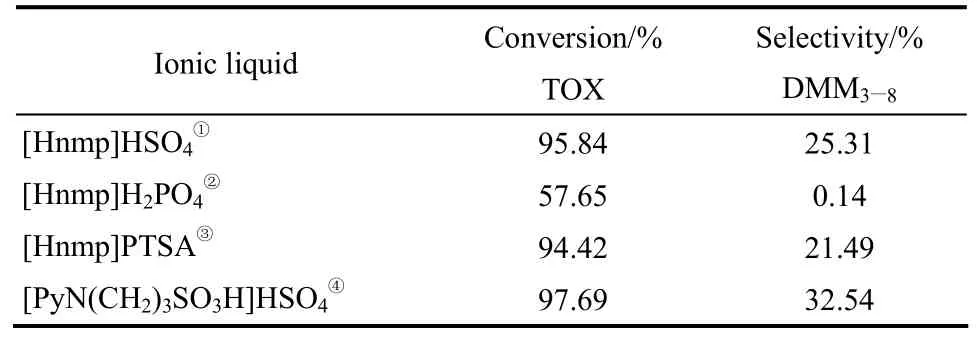
表9 不同離子液體的催化活性Table 9 Catalytic activities of various ionic liquids

Note:CH3OH /TOX=2:1 (molar ratio),110℃,6 h,2.0 MPa,catalyst amount=2.0%(mass).
2014年,張朝峰等[79]采用鍵合法將吡啶甲磺酸鹽離子液體負載到HZSM-5分子篩上,考察了分子篩負載型離子液體催化甲醇和三聚甲醛的縮醛化反應。在離子液體負載量為0.25 g/4 g HZSM-5、催化劑用量為 2.2%(質量分數)、CH3OH:TOX=1.5:1(摩爾比)、110℃的條件下反應3 h,DMM3—8的收率達到了67.35%。其催化活性好于單純使用離子液體或分子篩時的催化活性。固載化離子液體易回收、可重復利用,當重復使用3次后,DMM3—8收率降低到45.62%。
2 縮醛化合成聚甲氧基二甲基醚反應的動力學研究
2012年,Burger等[80]以離子交換樹脂(Amberlyst 46)為催化劑,開展了甲縮醛和三聚甲醛縮醛化反應合成聚甲氧基二甲基醚的動力學試驗,在50~90℃的條件下反應,根據實驗數據建立了 van’t Hoff方程lnKj=aj+bj/(T/K),得到了平衡常數(表10)。分別采用均相動力學模型(pseudohomogeneous kinetic model)和吸附動力學模型(adsorption-based kinetic model)進行了模擬計算。在吸附模型中,將吸附過程和表面反應過程進行了區分,其表面反應過程中反應速率很快,所以吸附過程是決速步,且吸附動力學模型計算結果與實驗結果更為接近,能夠很好地擬合間歇合成DMMn的反應過程。
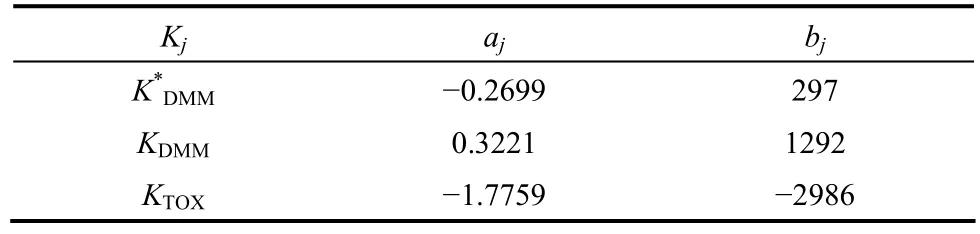
表10 ln Kj=aj + bj/(T/K)方程中參數的平衡常數Table 10 Parameters for bulk equilibrium ln Kj=aj + bj/(T/K)
劉奕等[81]采用硫酸、HZSM-5 分子篩、酸性陽離子交換樹脂等催化劑體系,根據串聯反應機理(圖13),只考慮甲醇濃度變化對反應的影響,假定各步串聯反應的反應級數相同,通過分析甲醇濃度隨時間變化數據,以假定的函數關系作圖得到該反應為一級反應。建立了反應動力學模型,依據Arrhenius方程,計算得到反應活化能Ea1~Ea5依次為79.52、79.97、95.69、108.10、120.38 kJ·mol-1。通過測量在不同溫度下的表觀反應速率,得到了反應動力學方程[式(2)~式(6)]。
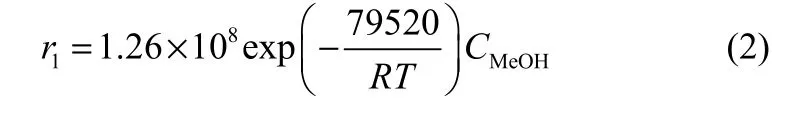

圖13 串聯反應合成DMMnFig.13 Propagation reactions of DMMn compounds

式中,rn依次為動力學模型中各步反應速率,mol·L-1·min-1。
Zhang等[82]以強酸性陽離子交換樹脂為催化劑,在固定床反應器上開展了甲醇和甲醛連續縮醛化反應合成DMMn的動力學實驗。認為對反應所使用的樹脂催化劑 HD-S(粒徑為 0.18~0.25 mm)而言,生成DMM1—4的小分子反應為一級反應,而生成大分子DMM5—6的反應則被認為是在催化劑表面的零級反應,在此基礎上建立了反應速率方程[式(7)~式(13)],并對反應速率方程進行計算,得到反應速率常數為 59278.38~130.19,表觀活化能為 38.58~43.68 kJ·mol-1(表 11)。類似地,張建強等[83]在固定床反應器中對甲縮醛和三聚甲醛縮醛化反應速率方程進行研究,根據連串反應機理建立了冪函數反應速率方程。對反應速率方程進行計算,得到了反應速率常數K1~K6依次為871.97、446.85、315.77、266.57、134.66、1.18。對反應速率方程參數進行了擬合回歸,反應速率方程較好地反映了甲縮醛和三聚甲醛的轉化率和產物分布。
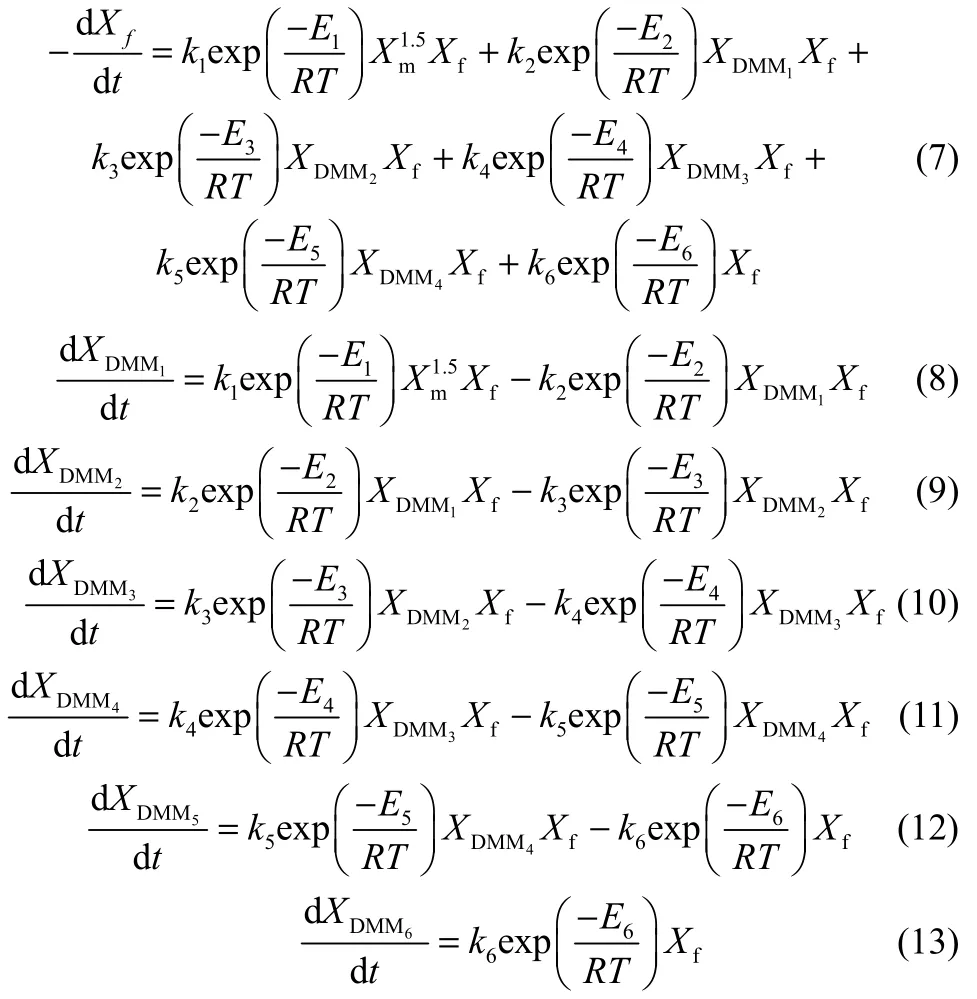
2015年,Zheng等[84]以離子交換樹脂 NKC-9為催化劑,開展了多聚甲醛和甲縮醛的縮醛化反應動力學實驗,研究發現反應液中DMMn產物分布遵循 Schulz-Flory分布模型,據此建立了反應速率矩陣[式(14)],通過計算得到了不同溫度下(60、70、80℃)正反應速率常數(kp)為 11.738、17.693、26.339,反應遵循二級反應動力學。逆反應速率常數(kd)為 0.037、0.065、0.108,反應遵循一級反應動力學。根據實驗數據計算了不同溫度下Kn的平均值為313.9、271.0、243.2,其值與n無關,與kp和kd有關。當催化劑量為5%(質量分數),正逆反應的指前因子分別為 1.84×107、5.36×106L·mol-1· min-1,正反應的活化能Ep(39.52 kJ·mol-1)低于逆反應的活化能Ed(52.01 kJ·mol-1),說明可逆反應的正反應為放熱反應(表12、表 13)。
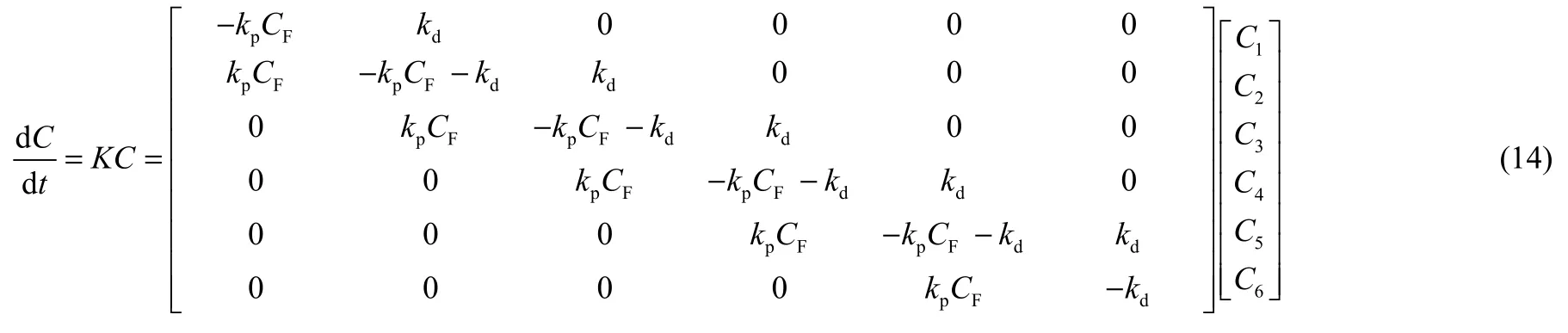

表11 酸性樹脂催化合成DMMn的動力學參數計算結果Table 11 Calculated results of kinetics parameters for DMMn synthesis over acid resin catalyst
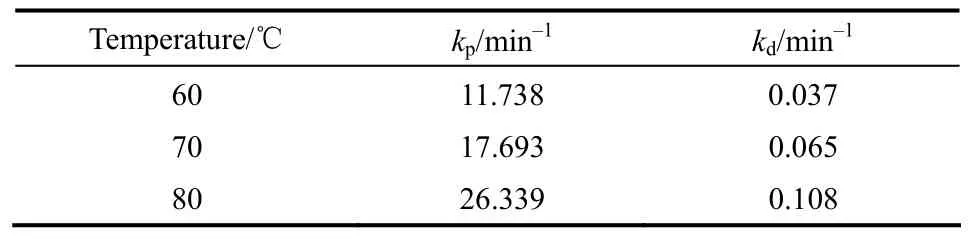
表12 鏈增長反應速率常數(kp)及解聚反應速率常數(kd)Table 12 Rate constants of propagation (kp) and depolymerization (kd) reactions of DMMn compounds respecting to 5%(mass) dosage of NKC-9 resin catalyst in designed space

表13 Arrhenius方程中的反應活化能(Ep, Ed)及指前因子(Ap, Ad)Table 13 Activation energy(Ep, Ed) and pre-exponential factor (Ap, Ad) of Arrhenius equation
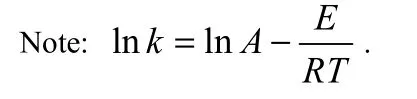
3 結論與展望
本文綜述了近年來縮醛化反應合成聚甲氧基二甲基醚催化劑體系、反應機理及動力學研究,通過典型的實例論述了液體酸、固體酸、離子液體等催化劑體系的研究進展。液體酸均相催化劑體系存在催化劑分離困難、對設備腐蝕嚴重、副產物多等缺點。固體酸催化劑體系中離子交換樹脂催化劑具有易分離、設備腐蝕性低等優點,但存在催化劑不耐高溫、酸性基團易流失等缺點。離子液體催化劑體系具有環境友好、酸性可調、對設備腐蝕低等優點,但存在催化劑價格昂貴等缺點。
盡管人們已在聚甲氧基二甲基醚合成及應用等領域取得了一些研究進展,但尚有很多亟待解決的問題,如對反應過程及機理研究較少,生產裝置的規模化與工程化,產品分離及微量雜質的控制,多醚類化合物應用領域的拓展等。基于以上問題,進一步開展聚甲氧基二甲基醚的高質化應用研究勢在必行。未來研究熱點可能會包括:根據不同的催化劑及反應體系,利用先進的表征手段加強對反應過程中作用機制的研究,為催化劑設計及產物分布控制提供依據;推進工業化應用進展,加強工程化及工藝放大研究力度;開展產品精細化分離及應用研究。
[1]VIGIER F,COUTANCEAU C,LEGER J M,et al.Polyoxymethylenedimethylether (CH3O(CH2O)nCH3) oxidation on Pt and Pt/Ru supported catalysts[J].J.Power Sources,2008,175(1):82-90.
[2]MOULTON D S,NAEGELI D W.Diesel fuel having improved qualities and method of forming:US5746785[P].1998-5-5.
[3]SANFILIPPO D,PATRINI R,MARCHIONNA M.Use of oxygenated product as a substitute of gas oil in diesel engines:EP1422285 B1[P].2004-5-26.
[4]SANFILIPPO D,PATRINI R,MARCHIONNA M.Use of an oxygenated product as a substitute of gas oil in diesel engines:US7235113 B2[P].2007-6-26.
[5]TEBBEN G D,SCHELING H,STROEFER E,et al.Biodiesel fuel mixture containing polyoxymethylene dialkyl ether:US20080216390 A1[P].2008-9-11.
[6]PATRINI R,MARCHIONNA M.Liquid mixture consisting of diesel gas and oxygenated compounds:EP1070755 B1[P].2001-1-24.
[7]VLASENKO N V,KOCHKIN Y N.Direct single-stage conversion of synthesis gas to dimethoxymethane:influence of the sequence of metal introduction into Cu,Pd-zeolite catalysts on the degree of Cu and Pd reduction and catalyst acidity[J].Russ.J.Appl.Chem.,2003,76(10):1615-1619.
[8]YUAN Y Z,LIU H C,IMOTO H,et al.Performance and characterization of a new crystalline SbRe2O6catalyst for selective oxidation of methanol to methylal original research article[J].J.Catal.,2000,195(1):51-61.
[9]YUAN Y Z,IWASAWA Y.Performance and characterization of supported rhenium oxide catalysts for selective oxidation of methanol to methylal[J].J.Phys.Chem.B,2002,106(17):4441-4449.
[10]ZHANG Q D,TAN Y S,YANG C H,et al.Catalytic oxidation of dimethyl ether to dimethoxymethane over Cs modified H3PW12O40/SiO2catalysts[J].J.Nat.Gas.Chem.,2007,16(3):322-325.
[11]LIU H C,IGLESIA E.Selective one-step synthesis of dimethoxymethaneviamethanol or dimethyl ether oxidation on H3+nVnMo12-nPO40Keggin structures[J].J.Phys.Chem.B,2003,107(39):10840-10847.
[12]STAUDINGER H,LUTHY M.Uber die konstitution der poly-oxymethylene[J].Helv.Chim.Acta,1925,8(1):41-64.
[13]WILLIAN F,RICHARD E.Preparation of polyformals:US2449469[P].1948-9-14.
[14]STROEFER E,SCHELLING H,HASSE H,et al.Method for production of polyoxymethylene dimethyl ethers from trioxan and dialkylethers:EP1902009[P].2008-3-26.
[15]STROEFER E,SCHELLING H,HASSE H,et al.Method for producing of polyoxymethylene dimethyl ethers:CA2581502 A1[P].2006-5-4.
[16]SCHELLING H,STROEFER E,PINKOS R,et al.Method for producing of polyoxymethylene dimethyl ethers:WO2006045506 A1[P].2006-5-4.
[17]SCHELLING H,STROEFER E,PINKOS R,et al.Method for producing of polyoxymethylene dimethyl ethers:US20070260094 A1[P].2007-11-8.
[18]STROEFER E,HASSE H,BLAGOV S.Method for producing of polyoxymethylene dimethyl ethers from methanol and formaldehyde:US20080207954 A1[P].2008-8-28.
[19]ZHAO Y P,XU Z,CHENH,et al.Mechanism of chain propagation for the synthesis of polyoxymethylene dimethyl ethers[J].J.Energy Chem.,2013,22(6):833-836.
[20]HAGEN G P,SPANGLER M J.Preparation of polyoxymethylene dimethyl ethers by catalytic conversion of dimethyl ether with formaldehyde formed by oxydehydrogenation of dimethyl ether:US5959156[P].1999-9-28.
[21]HAGEN G P,SPANGLER M J.Preparation of polyoxymethylene dimethyl ethers by catalytic conversion of dimethyl ether with formaldehyde formed by oxydehydrogenation of methanol:US6160174[P].2000-12-12.
[22]HAGEN G P,SPANGLER M J.Preparation of polyoxymethylene dimethyl ethers by catalytic conversion of dimethyl ether with formaldehyde formed by dehydrogenation of dimethyl ether:US6160186[P].2000-12-12.
[23]HAGEN G P,SPANGLER M J.Preparation of polyoxymethylene dimethyl ethers by catalytic conversion of dimethyl ether with formaldehyde formed by oxidation of methanol:US6166266[P].2000-12-26.
[24]HAGEN G P,SPANGLER M J.Preparation of polyoxymethylene dimethyl ethers by acid-activated catalytic conversion of methanol with formaldehyde formed by oxydehydrogenation of dimethyl ether:US6265528[P].2001-7-24.
[25]HAGEN G P,SPANGLER M J.Preparation of polyoxymethylene dimethyl ethers by catalytic conversion of formaldehyde formed by oxidation of dimethyl ether:US6392102[P].2002-5-21.
[26]HAGEN G P,SPANGLER M J.Preparation of polyoxymethylene dimethyl ethers by acid-catalytic conversion of methanol with formaldehyde formed by dehydrogenation of methanol:US6437195[P].2002-8-20.
[27]ARVIDSON M,FAKLEY M E,SPENCER M S.Lithium halide-assisted formation of polyoxymethylene dimethyl ethers from dimethoxymethane and formaldehyde[J].J.Mol.Catal.,1987,41(3):391-393.
[28]CLARISSE D,LAURENT G,JEANLUC C,et al.Mixtures of symmetrical and unsymmetrical polyoxymethylene dialkyl ethers,used in fuel compositions based on hydrocarbon distillates,especially diesel fuel:FR2906815A1[P].2008-12-12.
[29]BURGER J,SIEGERT M,STROFER E,et al.Poly(oxymethylene)dimethyl ethers as components of tailored diesel fuel:properties,synthesis and purification concepts[J].Fuel,2010,89(11):3315-3319.
[30]劉現立,田恒水,王賀玲,等.甲醛低聚反應研究[J].廣東化工,2009,36:23.LIU X L,TIAN H S,WANG H L,et al.Study on polymerization of formaldehyde[J].Guangdong Chemical Industry,2009,36: 23.
[31]陳婷,王亮,陳群,等.大孔強酸性陽離子交換樹脂催化甲縮醛和三聚甲醛合成聚甲醛二甲醚的研究[J].離子交換與吸附,2012,28(5):456-462.CHEN T,WANG L,CHEN Q,et al.Synehesis of polyoxymethylene dimethyl ethers from dimethoxymethane and trioxymethylene with cation resin as catalyst[J].Ion Exchange and Adsorption,2012,28(5):456-462.
[32]ZHENG Y Y,TANG Q,WANG T F,et al.Synthesis of a green diesel fuel additive over cation resins[J].Chem.Eng.Technol.,2013,36(11):1951-1956.
[33]隆寬燕,王濤,田恒水,等.聚甲醛二甲醚的綠色合成工藝研究[J].天然氣化工,2016,41(5):1-5.LONG K Y,WANG T,TIAN H S,et al.Environment-friendly process for synthesis of polyoxymethylene dimethyl ethers[J].Natural Gas Chemical Industry,2016,41(5):1-5.
[34]施敏浩,劉殿華,趙光,等.甲醇和甲醛催化合成聚甲氧基二甲醚[J].化工學報,2013,64(3):931-935.SHI M H,LIU D H,ZHAO G,et al.Catalytic synthesis of polyoxymethylene dimethyl ethers from methanol and formaldehyde[J].CIESC Journal,2013,64(3):931-935.
[35]劉顯科,夏成良,張建強,等.甲縮醛和甲醛催化合成柴油添加劑聚甲氧基二甲醚[J].石油化工,2015,44(7):888-892.LIU X K,XIA C L,ZHANG J Q,et al.Synthesis of polyoxymethylene dimethyl ethers as fuel additives from methylal and formaldehyde[J].Petrochemical Technology,2015,44(7):888-892.
[36]時米東,何高銀,代方方,等.氯化鋅催化甲醇和甲醛合成聚甲氧基二甲醚[J].化工學報,2016,67(7):2824-2831.SHI M D,HE G Y,DAI F F,et al.Synthesis of polyoxymethylene dimethyl ethers from methanol and formaldehyde catalyzed by ZnCl2[J].CIESC Journal,2016,67(7):2824-2831.
[37]馮偉樑,李豐,高煥新,等.聚甲醛二甲醚的制備方法:101768058 A[P].2010-07-07.FENG W L,LI F,GAO H X,et al.The preparation method of polyoxymethylene dimethyl ether:101768058 A[P].2010-07-07.
[38]李豐,馮偉樑,高煥新,等.分子篩催化合成聚甲醛二甲醚的方法:102040491 A[P].2011-05-04.LI F,FENG W L,GAO H X,et al.The method of polyoxymethylene dimethyl ether by molecular sieve catalysis:102040491 A[P].2011-05-04.
[39]李豐,劉志成,高煥新,等.催化合成聚甲醛二甲醚的方法:102295539 A[P].2011-11-28.LI F,LIU Z C,GAO H X,et al.The method of catalytic synthesis of polyoxymethylene dimethyl ether:102295539 A[P].2011-11-28.
[40]ZHAO Q,WANG H,QIN Z F,et al.Synthesis of polyoxymethylene dimethyl ethers from methanol and trioxymethylene with molecular sieves as catalysts[J].J.Fuel Chem.Technol.,2011,39(12):918-923.
[41]曹健,朱華青,王輝,等.分子篩催化劑催化合成聚甲氧基二甲醚[J].燃料化學學報,2014,42(8):986-993.CAO J,ZHU H Q,WANG H,et al.Synthesis of polyoxymethylene dimethyl ethers over zeolite catalysts[J].Journal of Fuel Chemistry and Technology,2014,42(8):986-993.
[42]張向京,武朋濤,張云,等.HMCM-22 分子篩負載磷鎢酸催化合成聚甲醛二甲醚[J].化學反應工程與工藝,2014,30(2):140-144.ZHANG X J,WU P T,ZHANG Y,et al.Synthesis of polyoxymethylene dimethyl ethers with HMCM-22 zeolite loading phosphotungstic acid as catalyst[J].Chemical Reaction Engineering and Technology,2014,30(2):140-144.
[43]WU J B,ZHU H Q,WU Z W,et al.High Si/Al ratio HZSM-5 zeolite:an efficient catalyst for the synthesis of polyoxymethylene dimethyl ethers from dimethoxymethane and trioxymethylene[J].Green Chem.,2015,17: 2353-2357.
[44]高曉晨,楊為民,劉志成,等.HZSM-5 分子篩用于合成聚甲醛二甲基醚[J].催化學報,2012,33(8):1389-1394.GAO X C,YANG W M,LIU Z C,et al.Catalytic performance of HZSM-5 molecular sieve for synthesis of polyoxymethylene dimethyl ethers[J].Chinese Journal of Catalysis,2012,33(8):1389-1394.
[45]何欣,袁志慶,騰加偉.聚甲氧基甲縮醛的制備方法:103539645 A[P].2014-01-29.HE X,YUAN Z Q,TENG J W.The preparation method of polyoxymethylene dimethyl ether:103539645 A[P].2014-01-29.
[46]劉志成,許云風,高曉晨,等.聚甲醛二甲醚催化劑及其應用:104549443 A[P].2015-04-29.LIU Z C,XU Y F,GAO X C,et al.Catalyst for polyoxymethylene dimethyl ether and its application:104549443 A[P].2015-04-29.
[47]李豐,馮偉樑,高煥新,等.聚甲醛二甲醚的合成方法:101768057[P].2010-07-07.LI F,FENG W L,GAO H X,et al.The method for synthesizing polyoxymethylene dimethyl ethers:101768057[P].2010-07-07.
[48]洪正鵬,商紅巖.一種合成聚甲醛二甲基醚的方法:101898943 A[P].2010-12-01.HONG Z P,SHANG H Y.A method for synthesizing polyoxymethylene dimethyl ethers:101898943 A[P].2010-12-01.
[49]ZHANG J Q,FANG D Y,LIU D H.Evaluation of Zr-alumina in production of polyoxymethylene dimethyl ethers from methanol and formaldehyde:performance tests and kinetic investigations[J].Ind.Eng.Chem.Res.,2014,53:13589-13597.
[50]趙峰,李華舉,宋煥玲,等.三聚甲醛與甲醇在/Fe2O3固體超強酸上的開環縮合反應研究[J].天然氣化工,2013,38(1):1-6.ZHAO F,LI H J,SONG H L,et al.Ring-opening and condensation of trioxane with methanol over/Fe2O3solid superacid[J].Natural Gas Chemical Industry,2013,38(1):1-6.
[51]LI H J,SONG H L,CHEN L W,et al.Designed/Fe2O3-SiO2solid acids for polyoxymethylene dimethyl ethers synthesis:the acid sites control and reaction pathways[J].Appl.Catal.B:Environ.,2015,165:466-476.
[52]LI H J,SONG H L,ZHAO F,et al.Chemical equilibrium controlled synthesis of polyoxymethylene dimethyl ethers over sulfated titania[J].J.Energy Chem.,2015,24(2):239-244.
[53]沈儉一,趙宇培,徐錚,等.一種合成聚甲醛二甲基醚的方法:102775284A[P].2012-11-14.SHEN J Y,ZHAO Y P,XU Z,et al.A method for synthesis of polyoxymethylene dimethyl ether:102775284A[P].2012-11-14.
[54]高曉晨,楊為民,劉志成,等.聚甲醛二甲醚的合成方法:103880614 A[P].2014-06-25.GAO X C,YANG W M,LIU Z C,et al.The synthesis method of polyoxymethylene dimethyl:103880614 A[P].2014-06-25.
[55]王一萌,付文華,梁筱敏,等.一種聚甲醛二甲醚的合成方法:104177237 A[P].2014-12-03.WANG Y M,FU W H,LIANG X M,et al.A method for synthesis of polyoxymethylene dimethyl ether:104177237 A[P].2014-12-03.
[56]FU W H,LIANG X M,ZHANG H D,et al.Shape selectivity extending to ordered supermicroporous aluminosilicates[J].Chem.Commun.,2015,51(8):1449-1452.
[57]FANG X L,CHEN J,YE L M,et al.Efficient synthesis of poly(oxymethylene) dimethyl ethers over PVP-stabilized heteropolyacids through self-assembly[J].Sci.China Chem.,2015,58(1):131-138.
[58]王建國,王瑞義,吳志偉,等.聚甲氧基二甲醚的制備方法:104086380 A[P].2014-10-08.WANG J G,WANG R Y,WU Z W,et al.The preparation method of polyoxymethylene dimethyl ether:104086380 A[P].2014-10-08.
[59]WANG R Y,WU Z W,QIN Z F,et al.Graphene oxide:an effective acid catalyst for the synthesis of polyoxymethylene dimethyl ethers from methanol and trioxymethylene[J].Catal.Sci.Technol.,2016,6:993-997.
[60]陳靜,唐中華,夏春谷,等.聚甲氧基甲縮醛的制備方法:200710018474.9[P].2008-05-21.CHEN J,TANG Z H,XIA C G,et al.Method for preparing polymethoxymethylal:200710018474.9[P].2008-05-21.
[61]CHEN J,TANG Z H,XIA C G,et al.Method for preparing polymethoxymethylal:US 7560599B2[P].2009-07-14.
[62]CHEN J,TANG Z H,XIA C G,et al.Process for synthesizing trioxymethylene using ionic liquid:US7598402B2[P].2008-11-27.
[63]陳靜,唐中華,夏春谷,等.離子液體催化合成聚甲氧基甲縮醛的方法:101665414B[P].2010-03-10.CHEN J,TANG Z H,XIA C G,et al.Method for synthesizing polyoxymethylene dimethyl ethers by ionic liquid catalysis:101665414B[P].2010-03-10.
[64]CHEN J,SONG H Y,XIA C G,et al.Method for synthesizing polyoxymethylene dimethyl ethers by ionic liquid catalysis:US8344183 B2[P].2013-01-01.
[65]CHEN J,SONG H Y,XIA C G,et al.Method for synthesizing polyoxymethylene dimethyl ethers catalyzed by an ionic liquid:US8816131B2[P].2014-8-26.
[66]XIA C G,SONG H Y,CHEN J,et al.Method for preparing polyoxymethylene dimethyl ethers by acetalation reaction of formaldehyde with methanol:US8987521B2[P].2016-02-26.
[67]CHEN J,SONG H Y,XIA C G,et al.System and method for continuously producing polyoxymethylene dialkyl ethers:US 9067188B2[P].2016-02-26.
[68]XIA C G,SONG H Y,CHEN J,et al.System and method for continuously producing polyoxymethylene dimethyl ethers:US 9169186B2[P].2015-10-27.
[69]CHEN J,SONG H Y,XIA C G,et al.Reaction system and process for prepaing polymethoxy dimethyl ether:US 9168503B2[P].2016-02-26.
[70]夏春谷,宋河遠,陳靜,等.連續制備聚甲氧基二甲醚的反應系統和工藝方法:103772163B[P].2014-05-07.XIA C G,SONG H Y,CHEN J,et al.System and method for continuously producing polyoxymethylene dimethyl ethers:103772163B[P].2014-05-07.
[71]WANG F,ZHU G L,LI Z,et al.Mechanistic study for the formation of polyoxymethylene dimethyl ethers promoted by sulfonic acid-functionalized ionic liquids[J].J.Mol.Catal.A-Chem.,2015,408:228-236.
[72]WU Y J,LI Z,XIA C G.Silica-gel-supported dual acidic ionic liquids as efficient catalysts for the synthesis of polyoxymethylene dimethyl ethers[J].Ind.Eng.Chem.Res.,2016,55(7):1859-1865.
[73]趙變紅,劉康軍,張朝峰,等.聚甲醛二甲醚合成反應中離子液體催化性能的比較[J].化工學報,2013,64(S1):98-103.ZHAO B H,LIU K J,ZHANG C F,et al.Comparison of catalytic properties of ionic liquids in polyoxymethylene dimethyl ethers synthesis reaction[J].CIESC Journal,2013,64(S1): 98-103.
[74]WU Q,WANG M,HAO Y,et al.Synthesis of polyoxymethylene dimethyl ethers catalyzed by Br?nsted acid ionic liquids with alkanesulfonic acid groups[J].Ind.Eng.Chem.Res.,2014,53(42):16254-16260.
[75]WU Q,LIW J,WANG M,et al.Synthesis of polyoxymethylene dimethyl ethers from methylal and trioxane catalyzed by Br?nsted acid ionic liquids with different alkyl groups[J].RSC Adv.,2015,5(71):57968-57974.
[76]趙強,李為民,陳清林.Br?nsted酸性離子液體催化合成聚甲醛二甲醚的研究[J].燃料化學學報,2013,41(4):463-468.ZHAO Q,LI W M,CHEN Q L.Synthesis of polyoxymethylene dimethy ethers catalyzed by Br?nsted acid ionic liquids[J].Journal of Fuel Chemistry and Technology,2013,41(4):463-468.
[77]李為民,趙強,左同梅,等.酸功能化離子液體催化合成聚縮醛二甲醚[J].燃料化學學報,2014,42(4):501-506.LI W M,ZHAO Q,ZUO T M,et al.Synthesis of polyoxymethylene dimethy ethers catalyzed by acidic functionalized ionic liquids[J].Journal of Fuel Chemistry and Technology,2014,42(4):501-506.
[78]鄧小丹,曹祖賓,韓冬云,等.復合催化劑合成聚甲氧基二甲醚的工藝研究[J].化學試劑,2014,36(7):651-655.DENG X D,CAO Z B,HAN D Y,et al.Preparation of polyoxymethylene dimethyl ethers catalyzed by composite catalyst[J].Chemical Reagents,2014,36(7):651-655.
[79]張朝峰,張海新,陳樹偉,等.吡啶甲磺酸鹽離子液體的固載化及其在聚甲醛二甲醚合成中的催化作用[J].燃料化學學報,2014,42(5):609-615.ZHANG C F,ZHANG H X,CHEN S W,et al.Immobilization ofN-(3-sulfopropyl) pyridinium methanesulfonate ionic liquid and its catalytic performance in the synthesis of polyoxymethylene dimethyl ethers[J].Journal of Fuel Chemistry and Technology,2014,42(5):609-615.
[80]BURGER J,STROFER E,HASSE H.Chemical equilibrium and reaction kinetics of the heterogeneously catalyzed formation of poly(oxymethylene) dimethyl ethers from methylal and trioxane[J].Ind.Eng.Chem.Res.,2012,51(39):12751-12761.
[81]劉奕,高曉晨,高煥新,等.合成聚甲醛二甲醚的反應動力學[J].化學反應工程與工藝,2014,30(4):365-370.LIU Y,GAO X C,GAO H X,et al.Kinetics of synthesis of polyoxymethylene dimethyl ethers[J].Chemical Reaction Engineering and Technology,2014,30(4):365-370.
[82]ZHANG J Q,SHI M H,FANG D Y,et al.Reaction kinetics of the production of polyoxymethylene dimethyl ethers from methanol and formaldehyde with acid cation exchange resin catalyst[J].Reac.Kinet.Mech.Cat.,2014,113(2):459-470.
[83]張建強,唐斌,夏成良,等.甲縮醛與三聚甲醛合成聚甲氧基二甲醚反應速率研究[J].化學反應工程與工藝,2015,31(1):69-76.ZHANG J Q,TANG B,XIAN C L,et al.Kinetics of polyoxymethylene dimethyl ethers synthesis from methylal and trioxymethylene[J].Chemical Reaction Engineering and Technology,2015,31(1):69-76.
[84]ZHENG Y Y,TANG Q,WANG T F,et al.Kinetics of synthesis of polyoxymethylene dimethyl ethers from paraformaldehyde and dimethoxymethane catalyzed by ion-exchange resin[J].Chem.Eng.Sci.,2015,134: 758-766.
date:2017-06-01.
Prof.CHEN Jing,chenj@licp.cas.cn
supported by the National Natural Science Foundation of China (21473225).
Advances in acetalization synthesis of polyoxymethylene dimethyl ethers
JIN Fuxiang1,SONG Heyuan1,2,KANG Meirong1,XIA Chungu1,CHEN Jing1
(1State Key Laboratory of Oxo Synthesis and Selective Oxidation,Lanzhou Institute of Chemical Physics,Chinese Academy of Sciences,Lanzhou730000,Gansu,China;2University of Chinese Academy of Sciences,Beijing100049,China)
Polyoxymethylene dimethyl ether compounds have high oxygen contents and cetane numbers (CN),low condensation point and cold filtration point,are promising environmental protection diesel additive to improve diesel combustibility,enhance combustion efficiency,and reduce emission of NOxand carbon smog pollutants.In this review,latest achievement in catalyst system,reaction mechanism and synthesis kinetics of polyoxymethylene dimethyl ether by acetalization reaction was summarized.The catalyst systems included liquid acid,solid acid,and ionic liquid systems in literature.The reaction materials contained end-group (—CH3,—OCH3) provider such as methanol,methylal and dimethyl ether,which were downstream products of methanol,and chain segment (—CH2O—) provider such as 1,3,5-trioxane (TOX),paraformaldehyde (PF) or formaldehyde(FA).Synthesis kinetic models and parameters of polyoxymethylene dimethyl ethers were reviewed briefly.The acetalization and kinetics research are moving towards the direction that benefits large scale commercial production.
polyoxymethylene dimethyl ethers; catalyst; liquid acid; solid acid; molecular sieves; ionic liquid;kinetics
O 643.32; O 643.12; TQ 203.2
A
0438—1157(2017)12—4471—15
10.11949/j.issn.0438-1157.20170706
2017-06-01收到初稿,2017-09-01收到修改稿。
聯系人:陳靜。
金福祥(1984—),男,助理研究員。
國家自然科學基金項目(21473225)。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
