免焙燒制備WP/SiO2加氫脫硫催化劑
2017-12-26 01:19:31朱對虎遇治權黃瑞民王安杰
華中師范大學學報(自然科學版) 2017年6期
關鍵詞:催化劑
朱對虎 , 遇治權, 王 偉, 黃瑞民, 王安杰
(1.銀川能源學院 石油化工學院, 銀川 750105; 2.遼寧省高校石油化工技術與裝備重點實驗室, 遼寧 大連 116024)
免焙燒制備WP/SiO2加氫脫硫催化劑
朱對虎1*, 遇治權2, 王 偉1, 黃瑞民1, 王安杰2
(1.銀川能源學院 石油化工學院, 銀川 750105; 2.遼寧省高校石油化工技術與裝備重點實驗室, 遼寧 大連 116024)
采用等體積浸漬法制備焙燒和免焙燒的催化劑前驅體,用H2程序升溫還原法制備焙燒和免焙燒的WP/SiO2催化劑.對制備的催化劑進行XRD和N2物理吸附表征,并用質量分數為0.8%的二苯并噻吩/十氫萘溶液來考察催化劑的加氫脫硫反應活性.與焙燒的WP/SiO2(C-WP/SiO2)催化劑相比,NC-WP/SiO2具有較大的比表面積和孔容;反應后,兩種催化劑的比表面積和孔容均降低.在NC-WP/SiO2上,DBT的轉化率略低于C-WP/SiO2,但均隨著溫度的增加而升高;隨著溫度的升高,兩種催化劑加氫中間體的選擇性降低,直接脫硫產物BP的選擇性升高,說明增加溫度有助于含硫化合物的脫硫.340℃,C-WP/SiO2催化DBT的反應速率(0.33 mol·g-1·min-1)略高于NC-WP/SiO2(0.28 mol·g-1·min-1);在NC-WP/SiO2上,DBT主要以DDS路徑脫硫,而在C-WP/SiO2上,加氫活性較高.
磷化鎢; 加氫脫硫(HDS); 二苯并噻吩; 免焙燒
過渡金屬磷化物(如Ni2P、MoP、WP等)催化劑以其良好的活性和穩定性在加氫精制(加氫脫硫、加氫脫氮、加氫脫氧和氫解等)領域受到越來越多的關注[1-5].其中WP催化劑活性較高,而且穩定性較好[6].傳統方法采用H2程序升溫還原(H2-TPR)相應氧化物前驅體制得磷化物催化劑[1,6].氧化物前驅體則通過高溫焙燒金屬鹽和磷酸銨鹽制得,較高的焙燒溫度(600℃左右)和較長的焙燒時間(3~5 h)[7-8],不僅耗費能源,還會造成催化劑高溫團聚,降低其比表面積導致其活性下降.Sun等發現,采用免焙燒法可以制備具有良好反應活性及穩定性的Ni2P/SiO2催化劑[9].因此,筆者將免焙燒制備WP/SiO2催化劑,并以二苯并噻吩(DBT)為含硫模型化合物來考察其反應活性.
1 實驗部分
1.1 試劑
二苯并噻吩(DBT)為分析純(上海阿拉丁生化科技股份有限公司),十氫萘、偏鎢酸銨((NH4)6W12O39·xH2O)、磷酸氫二銨((NH4)2HPO4)、石英砂(20~40 目)均為分析純試劑(國藥集團化學試劑有限公司),白炭黑為工業純(沈陽化工試劑有限公司).
1.2 催化劑制備
1.2.1 催化劑前驅體的制備 采用等體積共浸漬法制備催化劑前驅體:室溫下將化學計量的(NH4)6W12O39·xH2O和(NH4)2HPO4共溶于去離子水中,形成浸漬液;在不斷攪拌下將浸漬液逐滴加入到載體上,繼續攪拌30 min后靜置過夜,在120℃下烘干12 h,制得未焙燒的催化劑前驅體A.為對比,將A在500℃焙燒3 h制得焙燒的催化劑前驅體B.
1.2.2 催化劑前驅體的還原 采用H2程序升溫還原法(H2-TPR)制備催化劑:將上述制得前驅體A和B壓片、破碎、篩分至20~40目后裝填于管式反應器中,調節H2流量為150 mL/min,按如下程序還原制備:20(190)400(10)400(100)500(120)500℃(其中括號中代表所用時間),降至室溫后用O2/Ar (0.5 vol%)鈍化2 h,取出備用.將制得的催化劑分別記為NC-WP/SiO2和C-WP/SiO2.
1.3 HDS反應活性評價
0.2 g催化劑裝填于內徑為8 mm的不銹鋼固定床反應器中,按催化劑制備條件程序升溫還原后降至300℃.加氫脫硫(HDS)反應條件為:總壓4.0 MPa,重時空速(WHSV)為26.7 h-1,氫/油體積比750,反應溫度300~360℃,反應原料為0.8%(質量分數)DBT的十氫萘溶液.采用HP-6890型氣相色譜儀測定有機產物組成,色譜柱為HP-5型毛細管柱(Agilent).
通過改變停留時間τ算得動力學參數,即在保持氫油比恒定的條件下,通過改變液體和氫氣進料流速來調變停留時間.τ的計算如下[10]:
τ=

(1)
其中,τ單位為g·min·mol-1.
DBT轉化率(x)由下式求得:
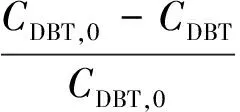
(2)
式中,CDBT,0為原料液中DBT濃度,CDBT為液體產物中DBT濃度.
各產物選擇性的計算如下:
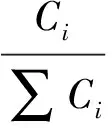
(3)
式中,Ci為產物i的濃度,∑Ci為產物中HDS產物濃度之和.
DBT進行HDS時遵循假一級反應方程如下:
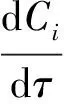
(4)
式中,k為假一級反應速率常數,x為DBT的轉化率.
1.4 催化劑表征
采用Rigaku D/Max 2400型X射線衍射儀表征催化劑的晶相,測定時采用Cu靶,Kα輻射,Ni濾波,管電壓40 kV,電流100 mA.采用Micrometritics Tristar II 3020在-196℃下測定催化劑的N2吸附等溫線,分別用BET和BJH理論計算其比表面積和孔容.
2 結果與討論
2.1 表征
圖1為采用H2-TPR制備的焙燒和免焙燒的WP/SiO2催化劑的XRD譜.可見,對于C-WP/SiO2和NC-WP/SiO2催化劑,均檢測出歸屬于WP的衍射峰,較寬的彌散峰歸屬于載SiO2.通過N2物理吸附測得NC-WP/SiO2的比表面積(138 m2·g-1)高于C-WP/SiO2催化劑(127 m2·g-1),這可能是由于高溫焙燒處理導致催化劑發生團聚,從而增大其顆粒尺寸,降低比表面積.而NC-WP/SiO2的孔容(0.58 cm3·g-1)低于C-WP/SiO2催化劑(0.61 cm3·g-1),可能由于未燒的無定形前驅體更易堵塞孔道.對于C-WP/SiO2和NC-WP/SiO2催化劑,進行HDS反應后催化劑的比表面積(分別由127和138 m2·g-1降至86和119 m2·g-1)和孔容(分別由0.61和0.58 cm3·g-1降至0.56和0.49 cm3·g-1)均降低(表1),可能由于反應后催化劑的孔道結構塌陷.
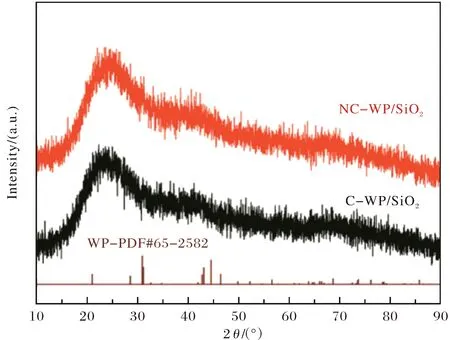
圖1 H2-TPR制備免焙燒和焙燒WP/SiO2的XRD譜Fig.1 XRD patterns of NC-WP/SiO2 and C-WP/SiO2 prepared by H2-TPR

catalystSBET/(m2·g-1)Vpore/(cm3·g-1)freshNC?WP/SiO2138058freshC?WP/SiO2127061spentNC?WP/SiO2119049spentC?WP/SiO286056
2.2 DBT的HDS反應活性
圖2為在C-WP/SiO2和NC-WP/SiO2催化劑上進行HDS時DBT轉化率隨溫度的變化.可以看出,DBT轉化率隨著反應溫度的升高而增加,但 C-WP/SiO2的催化活性略高于NC-WP/SiO2.圖式1示出DBT加氫脫硫的可能反應路徑,既可以通過直接脫硫(DDS)路徑形成聯苯(BP),也可以通過預加氫(HYD)路徑形成四氫和六氫中間體再發生C—S鍵斷裂形成苯基環己烷(CHB)和聯環己烷(BCH).通過對比產物的選擇性(圖3),可以看出在兩種催化劑上產物選擇性隨溫度的變化趨勢類似,低溫條件下,以HYD路徑產物為主;而在高溫條件下,以DDS路徑產物BP為主.
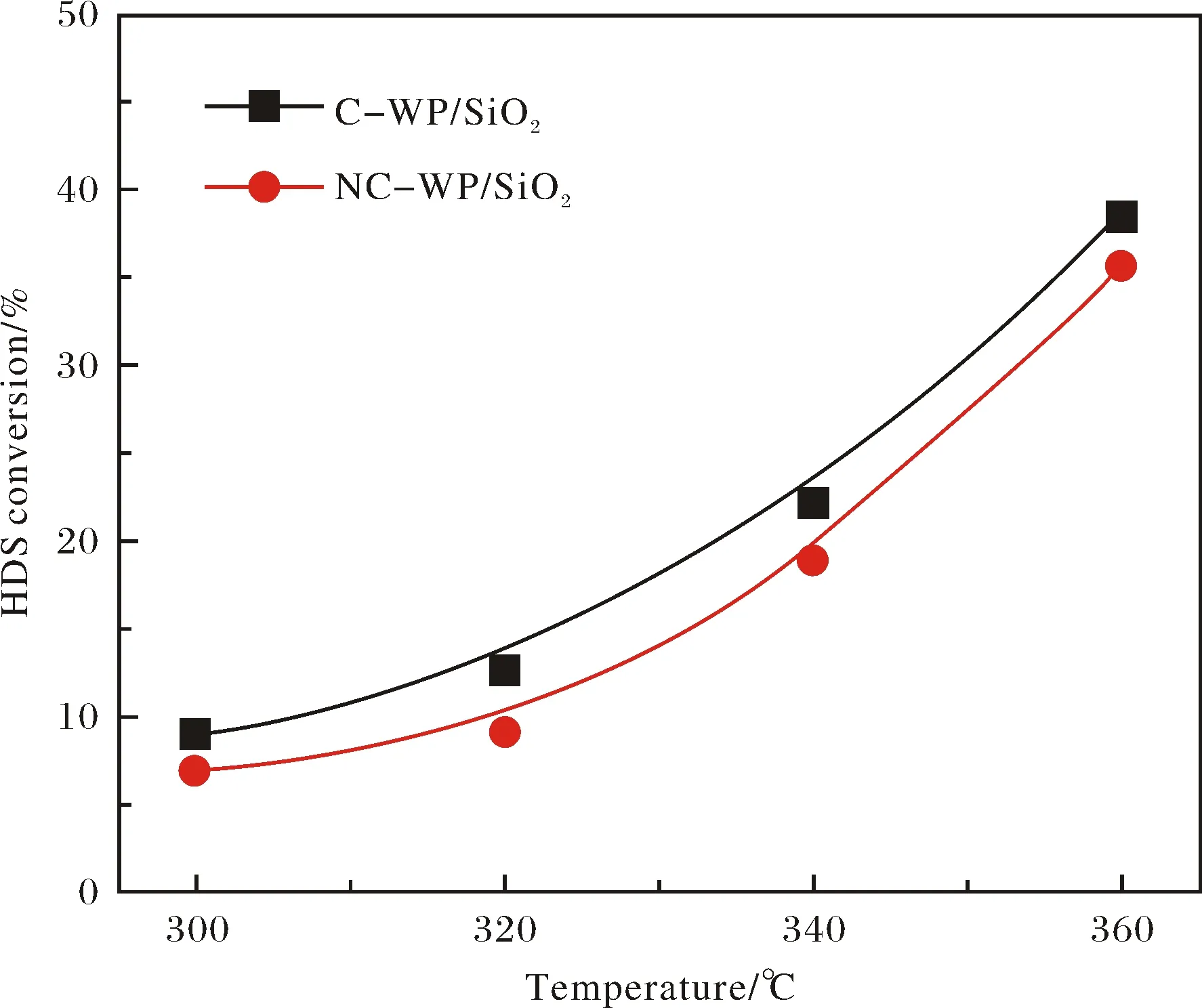
圖2 在C-WP/SiO2和NC-WP/SiO2催化劑上DBT轉化率隨溫度變化Fig.2 Variations of DBT conversion with temperature over C-WP/SiO2 and NC-WP/SiO2 catalysts
在340℃條件下,DBT在C-WP/SiO2和NC-WP/SiO2催化劑上進行HDS時DBT轉化率和產物選擇性隨停留時間τ的變化關系分別示于圖4和圖5.可以看出,在二種催化劑上,DBT轉化率隨著停留時間的增加而增加,且符合假一級動力學趨勢,但C-WP/SiO2的催化活性略高于NC-WP/SiO2.采用假一級反應模型計算得C-WP/SiO2和NC-WP/SiO2催化DBT的速率常數分別為0.33和0.28 mol·g-1·min-1,再次說明C-WP/SiO2的催化活性略高.DBT 在C-WP/SiO2和NC-WP/SiO2加氫脫硫的主要產物有DDS路徑產物BP,HYD路徑中間體TH-DBT和HH-DBT,以及HYD路徑終產物CHB和BCH.另外,還檢測到中間體CHEB-3的存在.相比于C-WP/SiO2催化劑,在NC-WP/SiO2上BP的選擇性較高,說明NC-WP/SiO2更有利于DDS路徑進行反應,而加氫活性略低;TH-DBT和HH-DBT的選擇性較高,而CHB和BCH的選擇性較低,同樣說明NC-WP/SiO2上DBT以HYD路徑進行脫硫的活性較低.
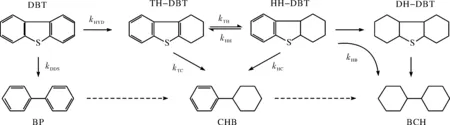
圖式1 DBT在WP/SiO2上的HDS反應網絡Scheme 1 Reaction network of DBT HDS over WP/SiO2
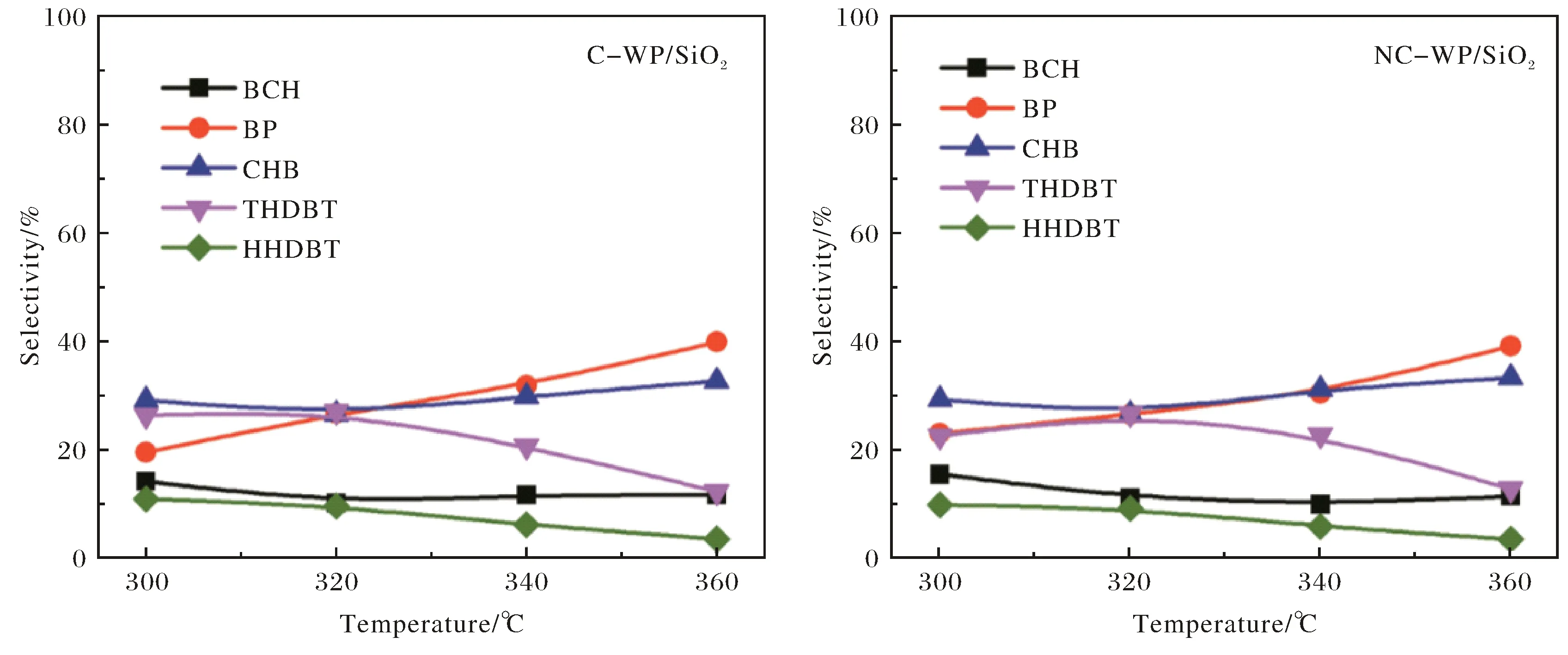
圖3 在C-WP/SiO2和NC-WP/SiO2催化劑上產物選擇性隨溫度變化Fig.3 Variations of product selectivities with temperature over C-WP/SiO2 and NC-WP/SiO2 catalysts
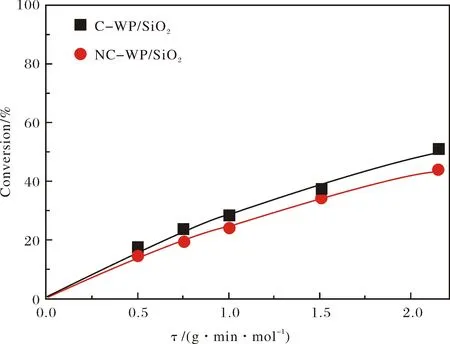
圖4 在340℃,C-WP/SiO2和NC-WP/SiO2催化劑上DBT轉化率隨停留時間變化Fig.4 Variations of DBT conversion with retention time at 340℃ over C-WP/SiO2 and NC-WP/SiO2 catalysts
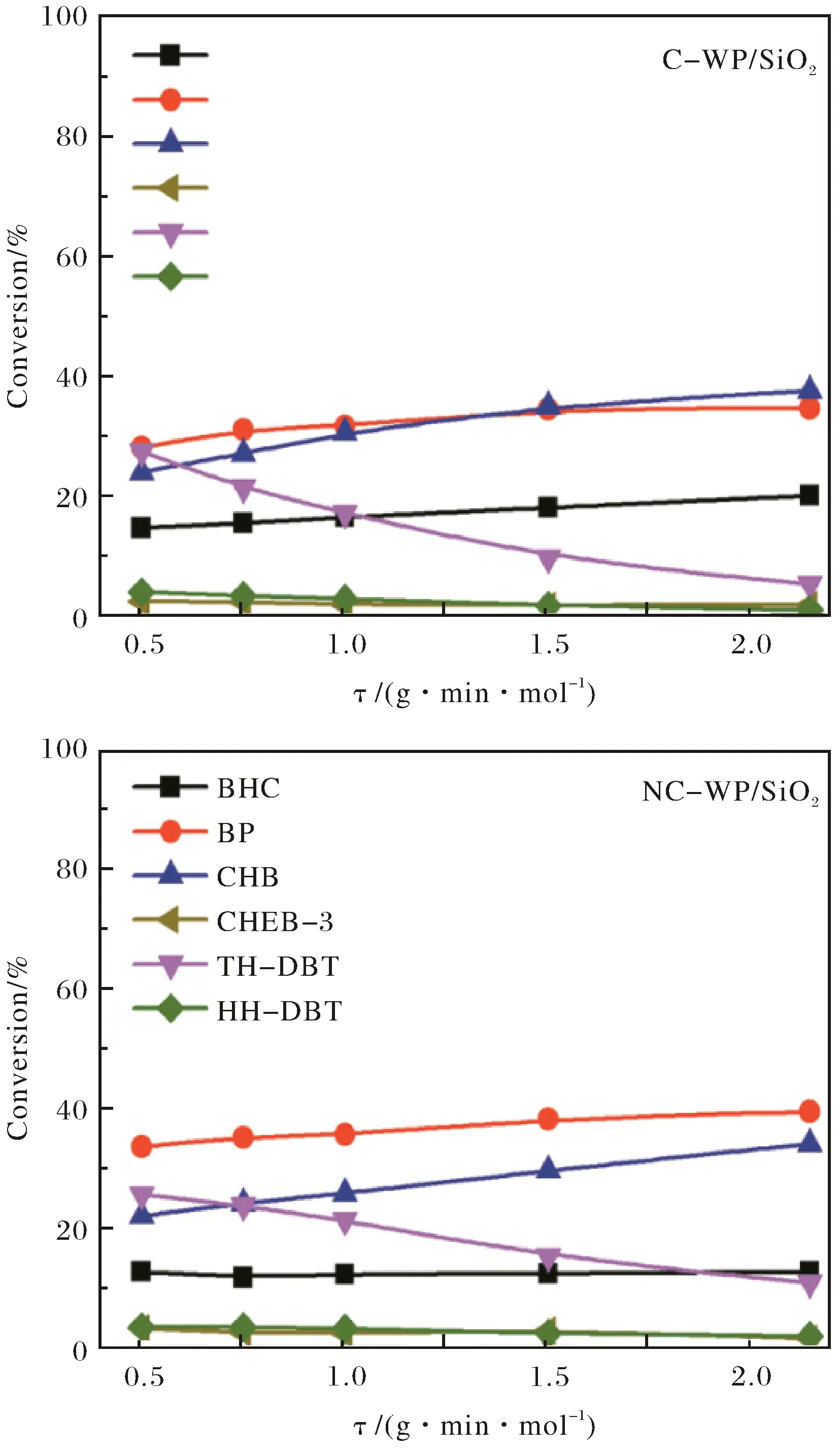
圖5 在340℃時C-WP/SiO2和NC-WP/SiO2催化劑上產物選擇性隨停留變化Fig.5 Variations of product selectivities with retention time at 340℃ over C-WP/SiO2 and NC-WP/SiO2 catalysts
3 結 論
1) 對于焙燒和免焙燒的催化劑前驅體,采用H2-TPR法均能成功合成出WP/SiO2催化劑;相較于C-WP/SiO2催化劑,NC-WP/SiO2具有較大的比表面積和略低的孔容;HDS反應后催化劑的比表面積和孔容均降低.
2) 相比于C-WP/SiO2催化劑,在NC-WP/SiO2催化劑上,DBT的轉化率略低,但變化趨勢一致,均隨著溫度的升高而增加;隨著溫度的升高,加氫中間體含量減少,促進脫硫反應的進行.
3) DBT在C-WP/SiO2和NC-WP/SiO2上進行HDS反應符合假一級動力學規律,前者的速率常數k(0.33 mol·g-1·min-1)略高于后者(0.28 mol·g-1·min-1);相比于C-WP/SiO2催化劑,在NC-WP/SiO2催化劑上,DBT更有利于通過DDS路徑進行脫硫,而加氫活性略低.
[1] OYAMA S T. Novel catalysts for advanced hydroprocessing: transition metal phosphides [J].Journal of Catalysis, 2003,216(1):343-352.
[2] LIU L H, LI G C, LIU D, et al.Preparation and catalytic performance of transition metal phosphides [J].Progress in Chemistry, 2010,22(9):1701-1708.
[3] WANG W, LI X, SUN Z C, et al.Influence of calcination and reduction methods on the preparation of Ni2P/SiO2and its hydrodenitrogenation performance [J].Applied Catalysis A: General, 2016,509(1):45-61.
[4] LIU P, CHANG W, WANG J, et al.MoP/Hβ catalyst prepared by low-temperature auto-combustion for hydroisomerization of n-heptane [J].Catalysis Communications, 2015,66(1):79-82.
[5] MA X, TIAN Y, HAO W, et al.Production of phenols from catalytic conversion of lignin over a tungsten phosphide catalyst [J].Applied Catalysis A: General, 2014,481(1):64-70.
[6] 孫福俠, 李 燦. 過渡金屬磷化物的加氫精制催化性能研究進展 [J]. 石油學報(石油加工), 2005,21(6):1-11.
[7] WANG R, SMITH K J. The effect of preparation conditions on the properties of high-surface area Ni2P catalysts [J]. Applied Catalysis A-General, 2010,380(1-2):149-164.
[8] GOTT T, OYAMA S T. A general method for determining the role of spectroscopically observed species in reaction mechanisms: Analysis of coverage transients (ACT) [J]. Journal of Catalysis, 2009,263(2):359-371.
[9] 孫志超, 王安杰, 李 翔, 等. 免焙燒制備Ni2P/SiO2加氫脫硫催化劑 [J]. 石油學報(石油加工), 2013,29(5):767-772.
[10] BAI J, LI X, WANG A, et al. Hydrodesulfurization of dibenzothiophene and its hydrogenated intermediates over bulk MoP [J]. Journal of Catalysis, 2012,287(1):161-169.
PreparationofWP/SiO2hydrodesulfurizationcatalystbyanon-calcinationmethod
ZHU Duihu1, YU Zhiquan2, WANG Wei1, HUANG Ruimin1, WANG Anjie2
(1.Yinchuan Institute of Energy, Institute of Petrochemical Technology, Yinchuan 750105, China; 2.Liaoning Key Laboratory of Petrochemical Technology and Equipment, Dalian,Liaoning 116024, China)
Calcinated and non-calcinated WP/SiO2precursors were prepared by an incipient impregnation method, followed by H2temperature-programmed reduction (H2-TPR) method. The morphologies of the catalysts were characterized by XRD and N2physical adsorption, and the HDS performances were studied with a model fuel containing 0.8% (mass fraction) dibenzothiophene (DBT) in decalin. Compared with the calcinated WP/SiO2(C-WP/SiO2), NC-WP/SiO2possessed larger specific surface area and pore volume. After HDS of DBT, the specific surface area and pore volume of both catalysts decreased. Over NC-WP/SiO2, the conversion of DBT was slightly lower than that over C-WP/SiO2. Over these two catalysts, the conversion of DBT increased with elevated temperature. Besides, as the reaction temperature increased, the selectivity of BP increased, while that of the hydrogenation intermediate decreased. It could be concluded that high temperature enhanced the HDS performance of the catalysts. At 340℃, C-WP/SiO2(0.33 mol·g-1·min-1) showed higherkthan that over NC-WP/SiO2(0.28 mol·g-1·min-1). Over NC-WP/SiO2, HDS of DBT mainly occurred through DDS pathway. On the other hand, the hydrogenation performance was much higher over C-WP/SiO2.
tungsten phosphide; hydrodesulfurization; DBT; non-calcination
2017-10-16.
寧夏高校科研專項基金資助項目(NGY2017246) ; 銀川能源學院重點項目(2016-KY-Z-11) .
*通訊聯系人. E-mail: 454948133@qq.com.
10.19603/j.cnki.1000-1190.2017.06.011
1000-1190(2017)06-0786-05
O643.38
A
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
