Bi2 WO6量子點(QDs)修飾Bi2MoO6-xF2x異質結的構筑及其催化活性增強機理
2018-01-04 21:18:44王丹軍申會東付夢溪
無機化學學報 2018年1期
關鍵詞:催化劑
王丹軍 申會東 付夢溪,2 王 嬋 郭 莉 楊 曉 付 峰
(1延安大學化學與化工學院,陜西省化學反應工程重點實驗室,延安 716000)
(2西安工業(yè)大學,材料與化工學院,西安 710021)
Bi2WO6量子點(QDs)修飾Bi2MoO6-xF2x異質結的構筑及其催化活性增強機理
王丹軍*,1申會東1付夢溪1,2王 嬋1郭 莉1楊 曉1付 峰1
(1延安大學化學與化工學院,陜西省化學反應工程重點實驗室,延安 716000)
(2西安工業(yè)大學,材料與化工學院,西安 710021)
采用簡單沉積-沉淀法合成了 Bi2WO6@Bi2MoO6-xF2x(BWO/BMO6-xF2x)異質結,借助 XRD、XPS、TEM、SEM、EDS、UV-Vis-DRS、PC和EIS等測試技術對其組成、形貌、光吸收特性和光電化學性能等進行系統(tǒng)表征,并以模型污染物羅丹明B(RhB)的光催化降解作為探針反應來評價Bi2WO6@Bi2MoO6-xF2x異質結的光催化活性增強機制。形貌分析表明,所得Bi2MoO6微球由大量厚度為20~50 nm的納米片組成;FE-SEM和HR-TEM分析表明,尺寸約為10 nm的Bi2WO6量子點均勻沉積在Bi2MoO6-xF2x微球表面,形成新穎的Bi2WO6@Bi2MoO6-xF2x異質結;與純Bi2MoO6或者Bi2WO6相比,1∶1Bi2WO6@Bi2MoO6-xF2x異質結表現(xiàn)出更好的光催化活性和光電流性質,其對RhB光催化降解的表觀速率常數(shù)分別為純BMO和BWO的6.4和11.6倍。PC和EIS圖譜分析表明,Bi2WO6量子點表面沉積顯著提高Bi2MoO6-xF2x光生電子/空穴的分離效率和遷移速率;活性物種捕獲實驗證明了·和h+是主要的活性物種。根據(jù)實驗結果,探討了F-摻雜和Bi2WO6量子點之間的協(xié)同效應對Bi2MoO6的光催化活性的影響機制。
沉積-沉淀法;量子點修飾;Bi2WO6/Bi2MoO6-xF2x異質結;光催化;協(xié)同效應
0 引 言
Bi基復合氧化物是一類新型多功能材料,廣泛應用于鐵電、壓電和非線性光學材料,其價帶由Bi6s軌道和O2p軌道雜化而成,二者的強相互作用使其具有較高的電荷流動性,從而具有光催化活性[1]。其中,Bi2MoO6是一種典型的Aurivillius結構化合物,由[Bi2O2]2+層與[MoO4]2-層交替出現(xiàn)堆積而成,禁帶寬度較窄(2.5~2.8 eV),能響應可見光而具有一定的光催化性能[2-5]。然而,Bi2MoO6具有光響應范圍較窄、光生電子/空穴對易于復合、且載流子的遷移速率緩慢等缺點,限制了其實際應用[6]。近年來,為了提高Bi基半導體的光催化性能,人們進行了各種各樣的改性研究,常見的有貴金屬沉積[7]、構筑異質結[8-9]和離子摻雜[10-12]等。其中,離子摻雜和表面修飾是拓寬Bi基半導體可見光吸收范圍、抑制光生載流子復合的最簡單而有效的途徑。
Zhu課題組[13]成功制備了 Bi2WO6-xF2x光催化劑,F(xiàn)元素摻雜有效地抑制了光生電子/空穴對的復合,從而顯著提高了Bi2WO6的光催化活性。Li等[14]制備了F摻雜BiVO4光催化劑,同樣表現(xiàn)出優(yōu)異的光催化活性。Chen課題組通過調節(jié)Ce的摻雜濃度調控Bi2MoO6的能帶結構和缺陷的濃度,顯著提高了催化劑活性[10]。此外,采用半導體耦合也可有效抑制光生載流子的復合,成為提高光催化劑性能的又一種有效策略。Yang等利用自組裝策略制備了Bi2WO6/氧化石墨烯復合催化材料,顯著提高了Bi2WO6對雙酚A的光催化降解活性和苯甲醇的選擇性氧化活性[15]。Liang課題組成功構筑了系列窄帶隙量子點修飾的Bi基半導體復合光催化材料(Cu2O/Bi2WO6[16],Cu2O/BiOBr[17]和 Cu2O/BiOBr[18]),實驗表明窄帶隙量子點表面修飾可以顯著拓寬Bi基光催化材料的可見光吸收效率,同時可提高光生電荷的分離效率[16-18]。近期,Liu課題組以TiO2納米帶為模板,成功構筑了的Bi2MoO6納米片和TiO2納米帶組裝而成的三維結構異質結構[19],Bi2MoO6和TiO2組成的異質結構具有優(yōu)越的寬譜響應活性,實驗結果表明Bi2MoO6/TiO2表現(xiàn)出優(yōu)異的光催化有機物降解活性和分解水產氧活性,且催化劑的穩(wěn)定性較好。Zhang課題組報道[20],Bi2MoO6/Bi2WO6復合物的光催化性能明顯優(yōu)于單組分的Bi2MoO6和Bi2WO6催化劑,這是由于Bi2MoO6和Bi2WO6二者形成異質結構有利于延長光生載流子的壽命,從而有利于提高催化劑的活性。
對元素摻雜的Bi基催化材料進行表面半導體耦合構筑異質結構,將元素摻雜和半導體表面耦合兩種改性手段結合,利用二者的協(xié)同作用,有利于更大程度地改進Bi基催化材料的催化性能。近期,Tan等[21]報道了一種新穎的Bi2O3/Bi2WO6-xF2x異質結構光催化劑,F(xiàn)離子和Bi2O3之間的協(xié)同效應可以有效的促進光生電子和空穴的分離和遷移,進而導致光催化活性明顯增強。然而,將摻雜與表面修飾同時用于Bi2MoO6基催化材料改性過程,尚未見文獻報道。
本文中,我們以Bi(NO3)3、NaF和Na2MoO4為起始原料,采用醇熱法合成了三維分級結構的Bi2MoO6-xF2x微球。在此基礎上,采用沉積-沉淀法將Bi2WO6量子點沉積在Bi2MoO6-xF2x表面,獲得新穎的Bi2WO6@Bi2MoO6-xF2x異質結。以模型染料羅丹明B(RhB)的降解為探針反應,初步探討了F離子摻雜和Bi2WO6量子點表面沉積對Bi2MoO6光催化活性的影響,結合UV-Vis-DRS、PC測試測試結果和自由基捕獲實驗,對Bi2WO6@Bi2MoO6-xF2x異質結的催化性能的增強機制進行了探討。
1 實驗部分
1.1 光催化劑的制備
1.1.1 Bi2MoO6-xF2x(BMO6-xF2x)的合成
將0.630 6 g分析純的 Bi(NO3)3·5H2O溶于13 mL乙二醇中,接著加入0.157 3 g的 Na2MoO4·2H2O,繼續(xù)攪拌至固體完全溶解,然后向上述溶液中滴加32.5 mL NH4F(0.013 2g)的無水乙醇溶液,繼續(xù)攪拌1 h,停止攪拌后將此混合溶液轉移到聚四氟乙烯不銹鋼高壓反應釜,于160℃反應12 h。待反應結束后,取出反應釜自然冷卻到室溫,將產生的沉淀依次進行水洗、醇洗6次,干燥、研磨得到BMO6-xF2x,所得樣品摻雜 F 的理論值為 55%(nF∶nMo),采用與制備BMO6-xF2x同樣的方法來合成純Bi2MoO6(BMO)樣品。
1.1.2 Bi2WO6/Bi2MoO6-xF2x(BWO/BMO6-xF2x)復合型催化劑的制備
準確稱取一定量(0.776 2,0.970 0,1.164 2 g)的Bi(NO3)3和0.610 0 g制備的 BMO6-xF2x,磁力攪拌 1 h將其分散于26 mL乙二醇中,得到懸浮濁液A。稱取一定量(0.264 0,0.330 0,0.396 0 g)的 Na2WO6溶于26 mL無水乙醇,攪拌至固體完全溶解,得到B溶液。隨后將B溶液滴加到A溶液中,在室溫下繼續(xù)攪拌1 h,最后將混合溶液移入到聚四氟乙烯不銹鋼高壓反應釜,在160℃的溫度下反應12 h。待反應結束后,自然冷卻至室溫離心分離所得沉淀,依次經離心、水洗、醇洗、干燥得粉末樣品,獲得Bi2WO6與 Bi2MoO6-xF2x物質的量之比為 0.8∶1,1∶1 和1.2∶1 的系列異質結,分別記作 0.8∶1BWO/BMO6-xF2x,1∶1BWO/BMO6-xF2x和 1.2∶1BWO/BMO6-xF2x。 采用同樣的方法,制備了純Bi2WO6(BWO)和1∶1BWO/BMO催化劑。
1.2 催化劑的表征
樣品的物相組成在日本島津XRD-7000型X射線粉末衍射儀上測定,Cu Kα(Ni濾玻片,λ=0.154 18 nm),管電壓 40 kV,管電流 30 mA,2θ=10°~80°;樣品的光電子能譜在美國PE公司PH5400型光電子能譜儀上檢測,鋁靶Al Kα 1 486.68 eV,激發(fā)功率250 W,用C1s電子結合能284.6 eV進行誤差校正;樣品的形貌在日本電子JEOL-6701型場發(fā)射掃描電子顯微鏡(FE-SEM)上觀察;樣品的高分辨透射電鏡(HR-TEM)在日本電子JEM-2100型透射電鏡上測試,加速電壓200 kV;樣品紫外-可見吸收光譜在日本島津UV-2550型紫外-可見分光光度計上測定,BaSO4作參比,掃描范圍200~800 nm。光電流和阻抗測定在CHI660D電化學工作站上進行,以鉑電極、飽和甘汞電極和樣品分別作為對電極、參比電極和工作電極的三電極系統(tǒng)進行分析,用0.1 mol·L-1的Na2SO4溶液作為電解液。工作電極的制備過程:將10 mg研磨樣品超聲分散到2 mL無水乙醇溶液中,形成泥漿。然后將泥漿分散到ITO上,在60℃的條件下,煅燒6 h,最后得到工作電極。
1.3 活性評價
樣品的催化活性評價在XPA-7型旋轉式光化學反應儀中進行,以400 W金鹵燈為實驗光源(發(fā)射光譜:380~800 nm),實驗時單位面積光強度為0.45 W·cm-2,利用濾光片過濾掉420 nm以下的光。實驗時,先將200 mL濃度為10 mg·L-1的RhB溶液加入光催化反應器中,再加入200 mg催化劑(催化劑濃度為100 mg·L-1),黑暗中攪拌30 min達到吸附-脫附平衡后,開啟光源進行光催化降解反應。每隔一定時間取樣離心分離催化劑,取上清液測定紫外-可見吸收光譜和最大吸收波長處的吸光度,以此來評價催化劑的光催化活性。
2 結果與討論
2.1 XRD和XPS分析
圖1是純 BWO、 純 BMO、BMO6-xF2x和系列BWO/BMO6-xF2x異質結的XRD圖。純Bi2MoO6的衍射峰可以看出,各特征衍射峰的位置與正交晶系Bi2MoO6(PDF No.76-2388)相吻合。由圖1a可以看出,2θ角 位 于 28.3°,32.8°,32.9°,47.0°,47.1°,55.8°,58.8°,68.7°和 75.9°的衍射峰可歸屬于正交晶系Bi2WO6(PDF No.39-0256)的(131),(200),(260),(202),(331),(262),(400),(103)和(204)晶面。 由圖 1a 可以看出F離子摻雜和BWO的修飾沒有導致Bi2MoO6晶體結構的變化。然而,從圖1b看出當F離子摻雜以后Bi2MoO6晶體的特征峰輕微的向左發(fā)生移動,這是由于F離子取代晶格氧,改變了[Bi2O2]層和[MoO6]層之間作用力所致[20]。從圖1b還可以看出,在樣品BWO/BMO6-xF2x的XRD圖中,當BWO量子點沉積時,Bi2MoO6的(131)晶面對應的衍射峰發(fā)生移動,表明BWO和BMO6-xF2x之間形成異質結構[21-22]。
圖 2 是純 BMO,BMO6-xF2x和 BWO/BMO6-xF2x的XPS圖譜。從圖2a上半部分可以看出,樣品BMO中含有Bi、Mo、O和C元素;圖2a下半部分可以看出,樣品 BWO/BMO6-xF2x中含有 Bi、Mo、W、O 和 F元素,其中C元素是歸屬于儀器本身的C污染源。圖 2(b~f)分別為樣品純 BMO和 BWO/BMO6-xF2x的Bi4f,Mo3d,W4f,O1s和 F1s相應的高分辨率的 XPS對比譜。由圖2b可見,純BMO中159.0和164.4 eV的能譜峰分別對應于Bi4f7/2和Bi4f5/2,表明了Bi3+的存在[23],且比 BMO6-xF2x和 BWO/BMO6-xF2x中 Bi4f的特征峰位置向高結合能移動,表明F取代O對Bi的化學環(huán)境產生一定的影響;由圖2c可知,純Bi2MoO6中特征峰位于235.44和232.29 eV處,對應六價鉬的Mo3d5/2和Mo3d7/2,歸屬于MoO6八面體中Mo特征峰[24],與純Bi2MoO6相比較,BMO6-xF2x、Mo3d3/2和Mo3d5/2的結合能向高結合能方向移動,這可能是由于F部分取代[MoO6]八面體中的單元中的O原子,從而改變了Mo原子周圍的化學環(huán)境,形成了部分F-Mo-O鍵[13];此外,樣品BWO/Bi2MoO6-2x中Mo3d的特征峰進一步向高結合能方向移動,這是由于BWO量子點的沉積在Bi2MoO6-2x表面形成異質結構,導致表面層結構發(fā)生變化;由圖2d可以看出,W4f的特征結合能為37.2和35.1 eV,分別對應于六價鎢的W4f5/2和W4f7/2[25]。圖2e看出,BWO/BMO6-xF2x異質結表面存在2種不同氧物種,結合能為529.8和531.8 eV處的能譜峰對應于樣品的晶格氧和樣品表面吸附水的羥基氧[26],相對于純Bi2MoO6,BWO/BMO6-xF2x樣品中在高結合能位置531.8 eV處的擬合峰比例增加,說明F摻雜和BWO量子點沉積會給BMO表面引入更多的羥基。此外,圖2f出現(xiàn)了2個特征峰,結合能為680.7 eV歸屬于Bi4s,而結合能為684.0 eV歸屬于F1s[22]。
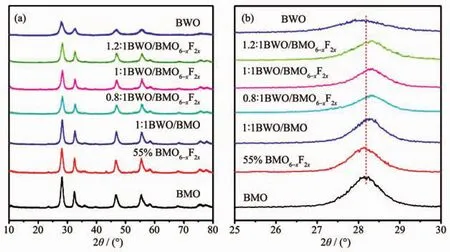
圖1 (a)純BWO、純BMO,BMO6-xF2x和系列BWO/BMO6-xF2x異質結XRD圖;(b)純BWO、純 BMO,BMO6-xF2x和系列 BWO/BMO6-xF2x異質結在 25°~30°的 XRDFig.1 (a)Pure BWO,pure BMO,BMO6-xF2xand series BWO/BMO6-xF2xheterojunction XRD spectra;(b)Enlargement of XRD patterns from 25°to 30°
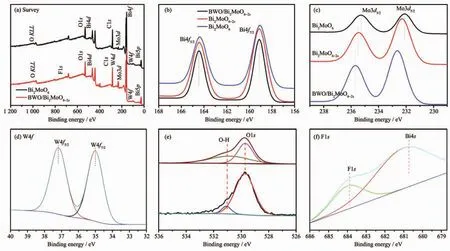
圖2 光催化劑Bi2MoO6,BMO6-xF2x和BWO/BMO6-xF2x的XPS圖譜Fig.2 XPS spectra of the Bi2MoO6,BMO6-xF2xand BWO/BMO6-xF2x:(a)Survey,(b)Bi4f,(c)Mo3d,(d)W4f,(e)O1s and(f)F1s
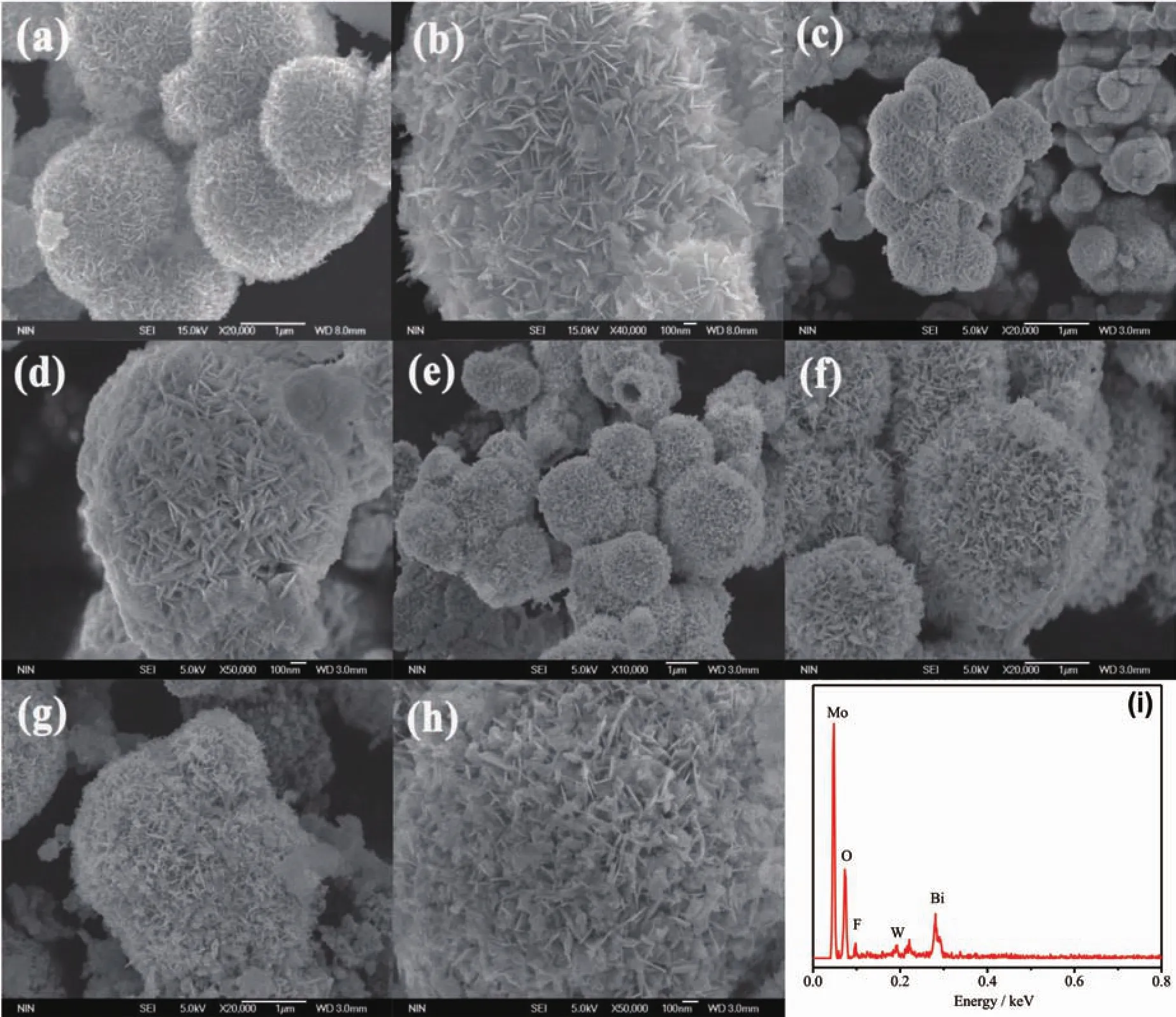
圖3 光催化劑的FE-SEM照片:(a,b)純BMO;(c,d)純BWO;(e,f)BMO6-xF2x;(g,h)BWO@BMO6-xF2x,(i)BWO/BMO6-xF2x的異質結EDS譜Fig.3 FE-SEM images of as-prepared(a,b)pure Bi2MoO6,(c,d)pure BWO,(e,f)BMO6-xF2xand(g,h)BWO/BMO6-xF2x heterostructures,(i)EDX spectrum of sample BWO/BMO6-xF2x
2.2FE-SEM和TEM分析
圖 3 是純 BMO、純 BWO、BMO6-xF2x和 1∶1BWO/BMO6-xF2x樣品的SEM照片,從圖3(a,b)可以看出,Bi2MoO6微球由大量的納米片組裝而成,平均直徑為 1~2 μm,而納米片的厚度為 20~50 nm。從圖 3(c,d)可以看出,BWO也是由納米片組裝而成的微球,平均直徑為 0.5~1 μm。 從圖 3(e,f)可以看出,BMO6-xF2x保持了Bi2MoO6微球結構,形貌沒有發(fā)生明顯的改變,表明F離子可能取代了BMO中的O原子。當BWO和BMO6-xF2x進行耦合,在組成BMO6-xF2x微球的納米片表面出現(xiàn)大量BWO顆粒,如圖3(g,h)。為了進一步確定在BMO表面BWO和F的存在,對BWO/BMO6-xF2x進行了EDX測試 (圖3i)。從圖 3i中可以看出,主要包含 Bi、Mo、W、O 和 F五種元素。圖4是BWO/BMO6-xF2x樣品的SEM圖像和元素映射圖像。圖 4(b~f)是 Bi、Mo、W、O 和 F 元素的分布圖像,其分布情況與EDX結果一致。圖5是BMO 和 1∶1BWO@BMO6-xF2x的 TEM/HR-TEM 照片,由圖5a可以清楚的看出Bi2MoO6是分級微球結構;圖5b是純BMO的HR-TEM照片,從圖中看出晶面間距為0.316 9 nm的晶面對應于BMO的 (131)晶面。圖5c是BWO@BMO6-xF2x的TEM照片,由圖可以清楚地發(fā)現(xiàn),BMO6-xF2x的表面有大量納米粒子;由圖5d可以清楚地看出,晶面間距為0.316 9 nm的晶面對應于BMO的(131)晶面,而晶面間距為0.315 0 nm的晶面則與BWO的(131)晶面相對應。
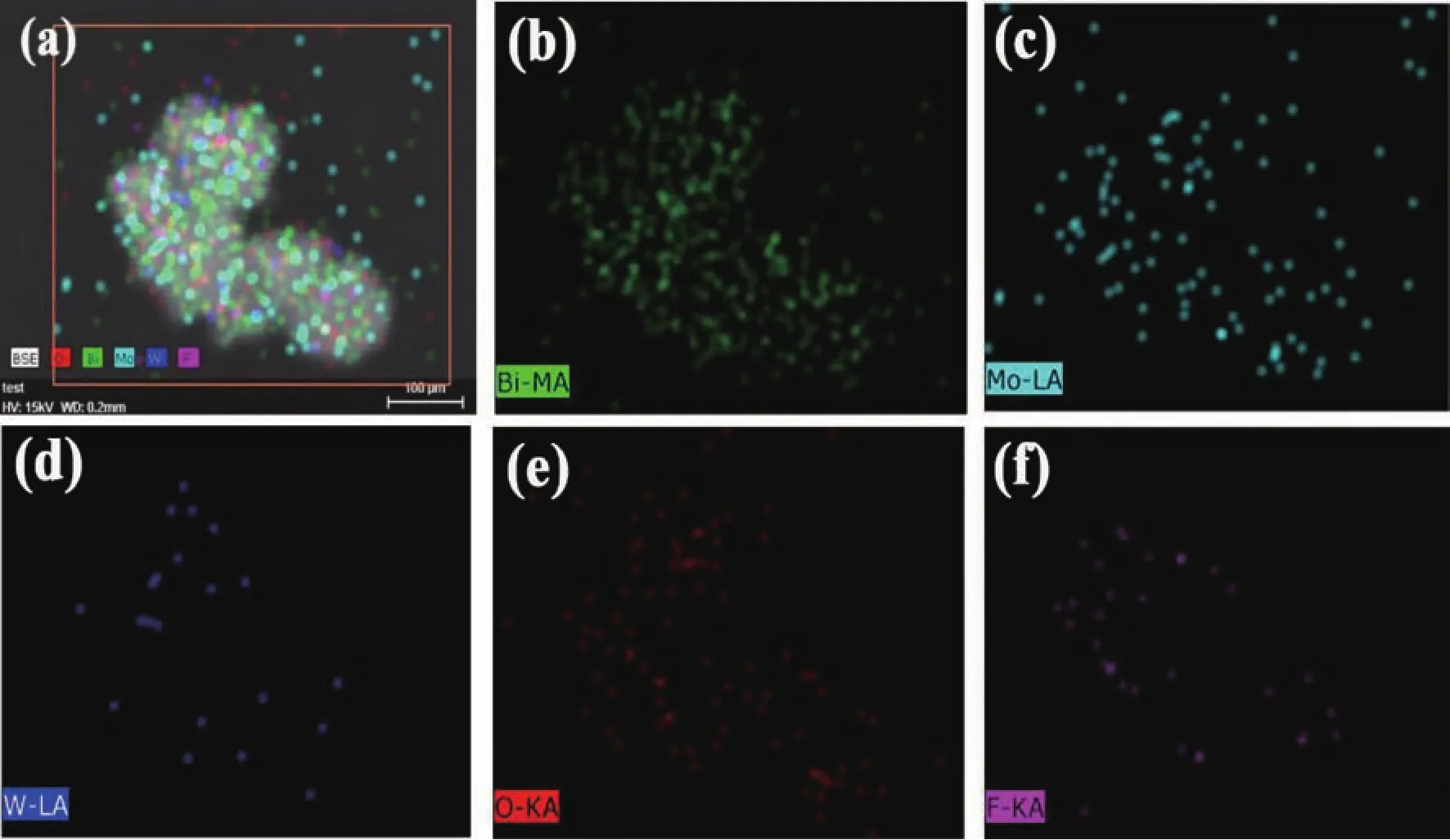
Fig.4 BWO@BMO6-xF2x樣品的SEM圖像和元素映射圖像Fig.4 SEM image and the elemental mapping images of BWO/BMO6-xF2x,(a)SEM image of BWO/BMO6-xF2x microsphere and the element mapping of(b)Bi,(c)Mo,(d)W,(e)O and(f)Felement
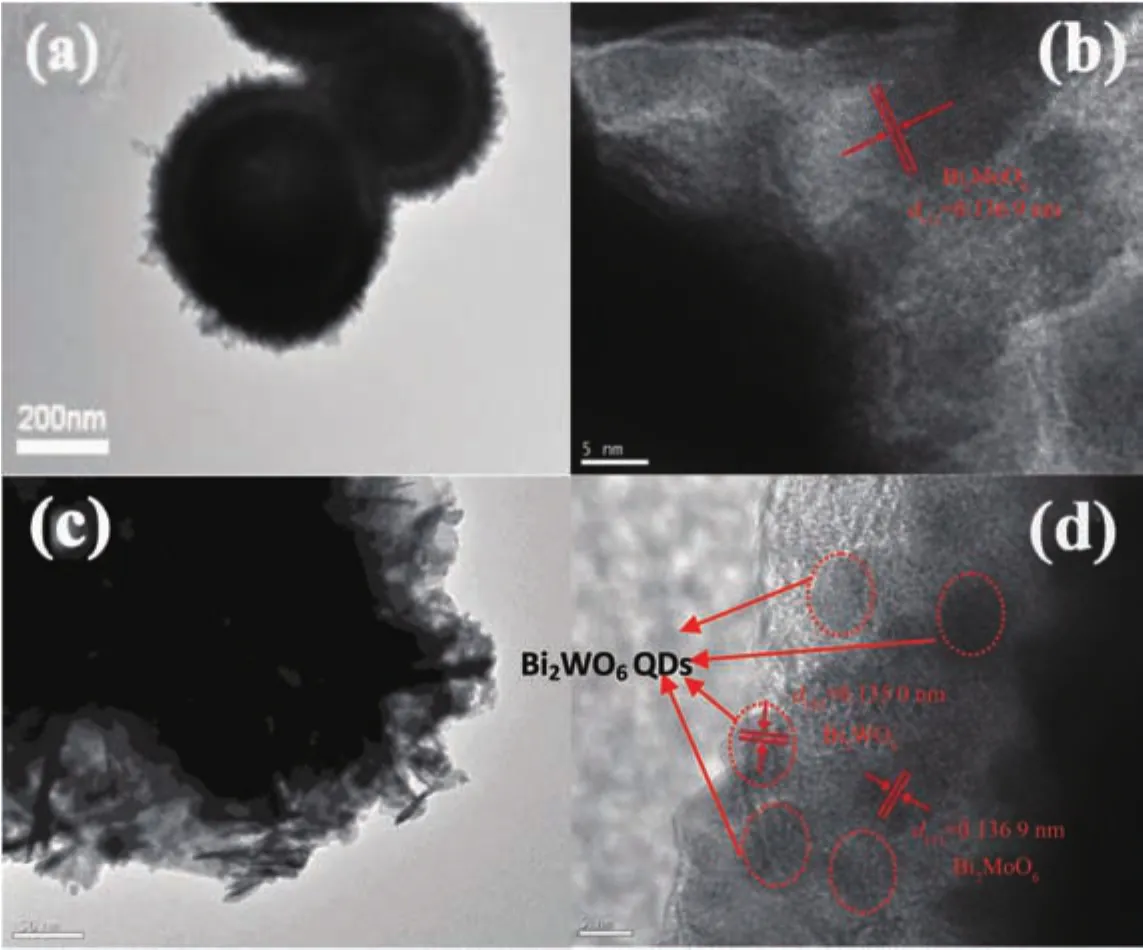
圖5 光催化劑的TEM/HR-TEM照片:(a)BMO;(c)BWO/BMO6-xF2x;(b)BMO and(d)1∶1BWO@BMO6-xF2x的高分辨透射電鏡照片F(xiàn)ig.5 TEM images of(a)BMO;(c)BWO/BMO6-xF2x;HR-TEM images of(b)BMO and(d)1∶1 BWO/BMO6-xF2x
2.3 光吸收特性
圖 6是純 BWO、 純 BMO、BMO6-xF2x和系列BWO/BMO6-xF2x異質結樣品的UV-Vis-DRS光譜。從圖可以看出,純Bi2MoO6和BWO的吸收邊分別在475和415 nm左右,當引入F離子后,Bi2MoO6的吸收邊發(fā)生紅移;當BWO和BMO6-xF2x耦合后,與BMO6-xF2x相比較,BWO/BMO6-xF2x異質結的吸收邊緣發(fā)生稍微的藍移,但二者建立界面電場利于光生電子/空穴對的分離和遷移,從而提高BMO6-xF2x的光催化活性。
春季露地櫻桃番茄栽培通常采用單桿整枝、改良單桿整枝或雙桿整枝。單桿整枝是生產上最常用的方式,它只保留主干,而將葉腋處長出的側枝全部摘除。其主要優(yōu)點是便于密植,一般每畝4000株,果實早期發(fā)育快,早期產量高,在生長期較短的地區(qū)或季節(jié)易于獲得高產。雙桿整枝是保留主干和第1花序下的第1側枝,其余側枝全部摘除,讓選留的側枝
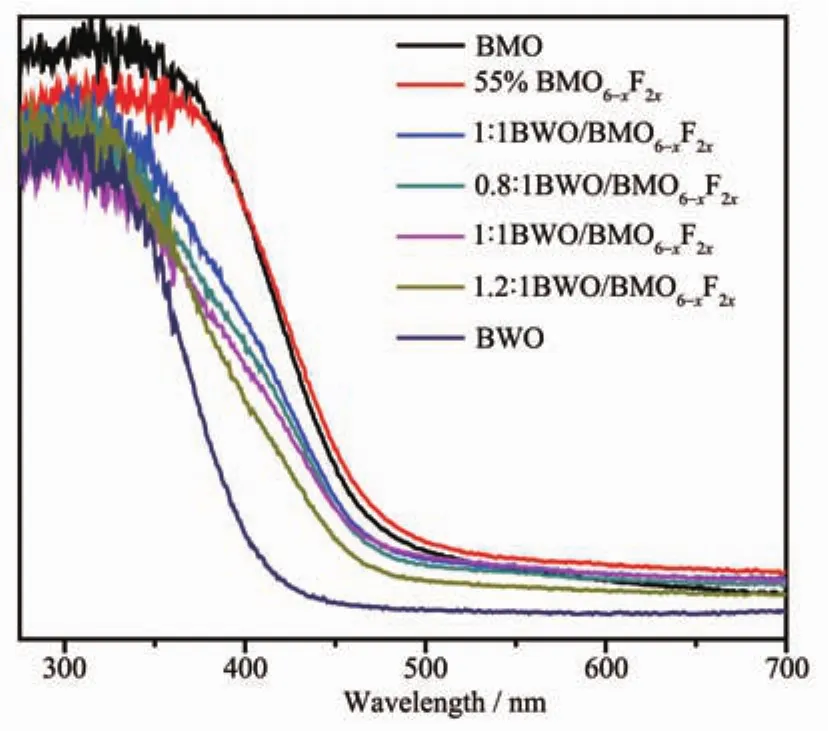
圖 6 純 BWO、BMO,BMO6-xF2x和系列 BWO/BMO6-xF2x異質結的UV-Vis吸收光譜Fig.6 UV-Vis absorption spectra of pure BWO,pure-BMO,BMO6-xF2xand series BWO/BMO6-xF2x complexes
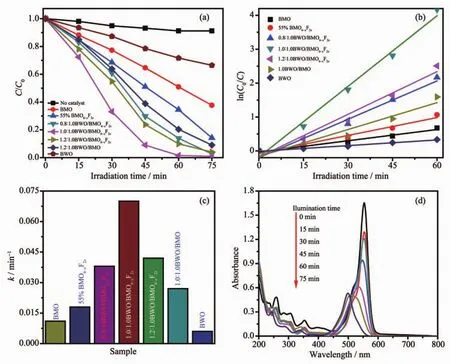
圖7 (a)不同催化劑可見光照射下降解RhB結果圖;(b)可見光照射下降解RhB的一級反應動力學圖譜;(c)光催化劑的光催化降解RhB的表觀速率常數(shù)對比圖;(d)1∶1BWO/BMO6-xF2x異質催化劑光催化降解羅丹明的吸收光譜隨時間的變動圖Fig.7 (a)Photocatalytic degradation efficiency of the different photocatalyst for RhB under visible light irradiation;(b)First-order kinetic fit for the degradation of RhB;(c)Apparent rate constant of different photocatalyst for RhB;(d)Change of the absorption spectra of photodegradated RhB with increasing irradiation time under visible light using 1∶1BWO/BMO6-xF2x composite as a photocatalyst
2.4 光催化活性分析
圖7為可見光照射下不同催化劑光催化降解模型污染物RhB的活性比較。從圖7a可以看出,BWO和BMO均對RhB有一定的降解活性,可見光照射60 min,RhB的降解率分別為29%和49%;F離子修飾BMO也表現(xiàn)出優(yōu)于單組份BMO的光催化活性。與BWO和BMO兩種純組分相比較,BWO/BMO6-xF2x異質結對RhB的降解速顯著增強,當nBWO∶nBMO6-xF2x的比值增加時,BWO/BMO6-xF2x異質結的活性增強,當二者比例達到1∶1時活性最高,而nBWO∶nBMO6-xF2x>1∶1 時活性反而下降;以 1∶1BWO@BMO6-xF2x異質結為催化劑可見光光照60 min后,RhB去除率接近100%。1∶1BWO/BMO6-xF2x活性最高的原因在于:BWO量子點沉積在BMO6-xF2x的表面,有利于提高BMO6-xF2x光生載流子的壽命,從而提高其催化活性,所以當 nBWO∶nBMO6-xF2x<1∶1 時活性隨著 BWO 沉積量的增加而增加;當BWO的沉積量大于1∶1時,由于過量的BWO占據(jù)了BMO6-xF2x光生表面的活性位點,抑制了電子在界面上的轉移,從而導致催化劑的活性反而降低[19]。
RhB染料降解滿足準一級動力學方程[27-28],圖7(b,c)給出了降解RhB的ln(C0/C)和照射時間圖譜和表觀速率常數(shù)(kapp)的值,從圖7c可以看出,BWO@BMO6-xF2x異質結對RhB降解的活性高于純BMO和BWO。此外,BWO/BMO6-xF2x異質結的催化活性與組成有關,1∶1BWO/BMO6-xF2x對RhB的光催化降解的速率常數(shù)最高,分別為純BMO和BWO的6.4和11.6倍。圖7d是光催化降解RhB的吸收光譜隨著輻照時間的變化圖譜。從圖中可以看出,在開始光降解的時候,RhB染料的最大吸光度的位置為554 nm,隨著輻照時間增加,吸收峰逐漸降低,并且發(fā)生藍移,直到輻照75 min后,吸收峰的位置為498 nm,這是因為在光催化降解RhB的過程中,發(fā)生了去甲基化和脫乙基[29]。
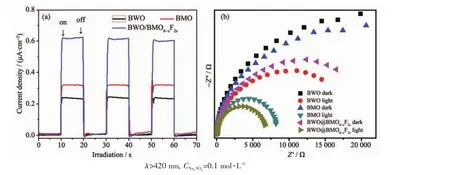
圖8 (a)光催化劑的瞬態(tài)光電流響應和(b)光催化劑的電化學阻抗圖譜Fig.8 (a)Comparison of transient photo-current responses of pure BMO,pure BWO and 1∶1BWO/BMO6-xF2xunder visible light irradiation;(b)Electrochemical impedance Nyqusit plots of pure BMO,pure BWO and 1∶1BWO/BMO6-xF2x heterojunction electrodes under dark and light irradiation
2.5 異質結光催化活性的增強機制
通常借助瞬態(tài)光電流響應譜來研究催化材料光生載流子的遷移和轉移過程。一般說來,高的光電流意味著電子和空穴的分離效率高,表現(xiàn)出較高的催化活性[30]。從圖8a中可以看出,與純BMO和BWO相比較,BWO@BMO6-xF2x異質結的瞬態(tài)光電流密度明顯增強,分別是BMO和BWO的2.5倍和1.9倍。因此,BWO/BMO6-xF2x異質結具有較高的光生載流子遷移和轉移速率。電化學阻抗譜(EIS)是另一種反映光生電子和空穴遷移和轉移過程有用的方法。眾所周知,在EIS譜中,弧的半徑反映了發(fā)生在電極表面的界面層電阻,弧的半徑越小意味著電荷轉移效率就越高[31-32]。從圖8b中可以看出,與純BMO和BWO相比較,BWO/BMO6-xF2x異質結催化劑的弧半徑最小,這表明BWO/BMO6-xF2x異質結具有更強的光生電子-空穴對轉移的能力,這表明F離子和BWO二者的協(xié)同作用可提高光生電子空穴對的分離能力,從而有助于提高光催化降解效率。
此外,為了確定光催化降解RhB過程中產生的主要活性物種,我們選擇乙二胺四乙酸二鈉(EDTA-2Na)、異丙醇(IPA)和苯醌(BQ)作為捕獲劑,分別來捕獲 h+、·OH 和·O2-[33-34]。從圖 9 上可以看出,IPA 的加入幾乎不影響1∶1BWO/BMO6-xF2x光催化降解RhB的效率,表明在光催化降解過程中,·OH不是反應的主要活性物種;當體系中存在BQ和EDTA-2Na時,RhB光催化降解均受到抑制,表明在RhB光催化降解過程中,h+和·O2-是主要的活性物種。
基于活性物種捕獲實驗結果,我們提出了1∶1BWO@BMO6-xF2x異質結光催化降解RhB的電荷轉移機理圖(圖 10)。 1∶1BWO@BMO6-xF2x異質結具有較高的光催化活性應該歸因于F離子和BWO的協(xié)同效應。一方面,F(xiàn)摻雜可在BMO的價帶頂部一些包含F(xiàn)2p和O2p新能級形成[35],導致BMO的吸收邊紅移,從而拓寬了可見光的利用范圍;另一方面,F(xiàn)-作為一種n型雜質元素,有利于增加BMO的電子密度,從而提高了光照射下BMO的光電流密度[36],進而提高BMO6-xF2x的光催化活性。在可見光照射下BMO6-xF2x和BWO均可被激發(fā)產生的光生電子/空穴對 (e-/h+),BMO6-xF2x導帶上的光生電子易于向BWO的導帶遷移,而BWO價帶上的空穴很容易遷移到BMO6-xF2x。此外,BWO量子點沉積在BMO6-xF2x的表面,二者建立界面電場利于光生電子/空穴對的分離和遷移,遷移至催化劑表面的光生電子被O2捕獲,形成具有氧化能力的,產生的和光生空穴作為主要的活性物種和RhB分子發(fā)生反應,使其分解為CO2,H2O等小分子[36-37]。從圖11可以看出,催化劑循環(huán)使用5次活性略有降低(圖11a),且使用前后催化劑的物相組成沒有發(fā)生變化(圖11b),表明了1∶1BWO@BMO6-xF2x比較穩(wěn)定。
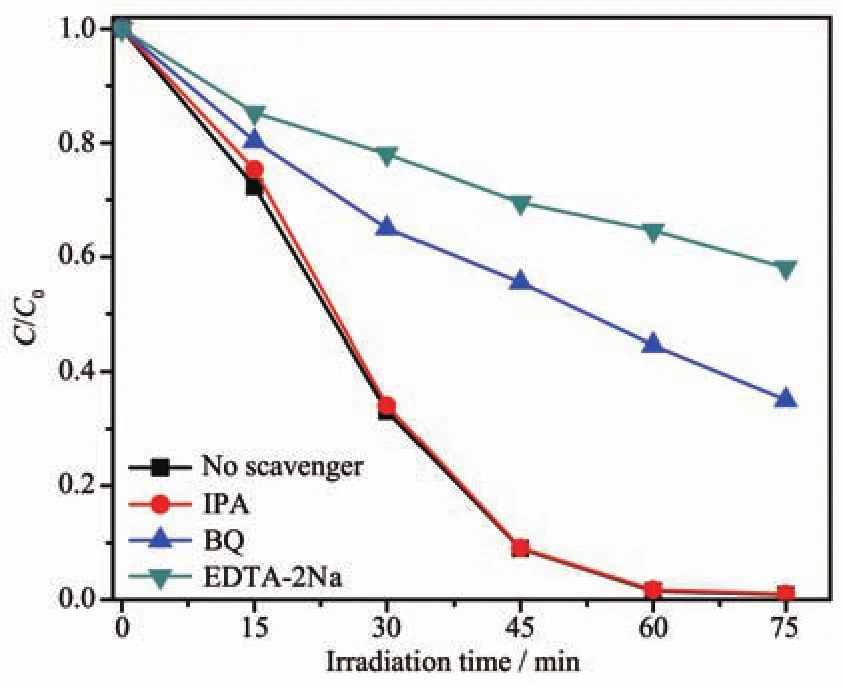
圖9 可見光照射下1∶1BWO/BMO6-xF2x光催化降解RhB的活性物種捕獲實驗Fig.9 Trapping experiments of active species in the system of photodegradation of RhB by 1∶1BWO@BMO6-xF2xunder visible light irradiation
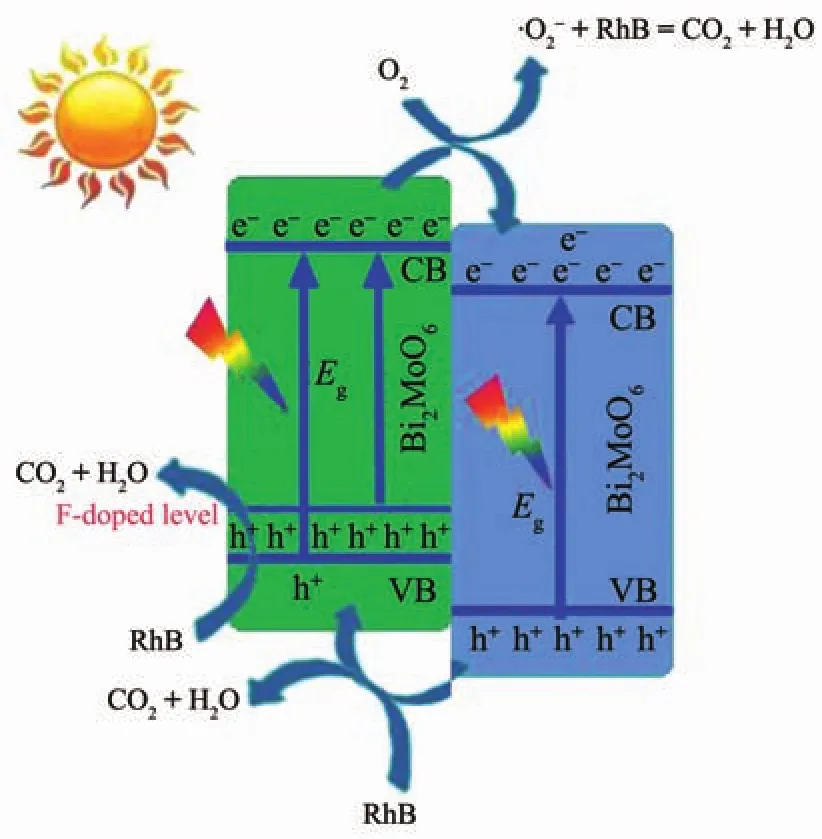
圖10 可見光照射下1∶1BWO@BMO6-xF2x異質結光催化降解RhB的電荷轉移機理圖Fig.10 Schematic illustration for the charge transfer echanism of rhodamine B in 1∶1BWO@BMO6-xF2xheterostructures under visible light irradiation
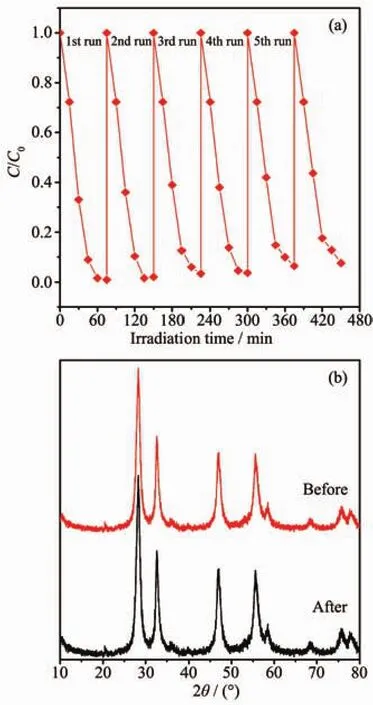
圖11 可見光照射下1∶1BWO@BMO6-xF2x異質結光催化降解RhB的穩(wěn)定性實驗Fig.11 Repeated experimental of photocatalytic degradation of RhB on 1∶1BWO@BMO6-xF2xheterostructures under visible light irradiation
3 結 論
(1)形貌分析表明,所得Bi2MoO6微球由大量厚度約20~50 nm的納米片組成;FE-SEM 和HR-TEM分析表明,尺寸約為10 nm的Bi2WO6量子點均勻沉積在Bi2MoO6-xF2x微球表面,形成的新穎的Bi2WO6/Bi2MoO6-xF2x異質結。
(2)實驗結果表明,異質結1∶1Bi2WO6@Bi2MoO6-xF2x的光催化活性最高,其對RhB光催化降解的表觀速率常數(shù)分別為純BMO和BWO的6.4和11.6倍。
(3)PC和EIS分析表明,Bi2WO6量子點沉積顯著提高了Bi2MoO6-xF2x光生電子/空穴的分離效率和遷移速率;活性物種捕獲實驗證明了·O2-和h+是主要的活性物種。
(4)根據(jù)實驗結果,F(xiàn)摻雜有利于增加Bi2MoO6的電子密度,Bi2WO6量子點與Bi2MoO6-xF2x光之間建立的界面電場利于光生電子/空穴對的分離,二者的協(xié)同效應導致Bi2MoO6的光催化活性顯著增強。
[1]Sun S M,Wang W Z.RSC Adv.,2014,4:47136-47152
[2]Shimodaira Y,Kato H,Kobayashi H,et al.J.Phys.Chem.B,2006,110:17790-17797
[3]Li H P,Deng Q H,Liu J Y,et al.Catal.Sci.Technol.,2014,4:1028-1037
[4]Jung J C,Kim H,Choi A S,et al.Catal.Commun.,2007,8:625-628
[5]Tian G H,Chen Y J,Zhou W,et al.J.Mater.Chem.,2011,21:887-892
[6]Yue D,Chen D M,Wang Z H,et al.Phys.Chem.Chem.Phys.,2014,16:26314-26321
[7]Mohamed R M,Ibrahim F M.J.Ind.Eng.Chem.,2015,22:28-33
[8]Zhang M Y,Shao C L,Mu J B,et al.CrystEngComm,2012,14:605-612
[9]Yan T,Yan Q,Wang X D,et al.Dalton Trans.,2015,44:1601-1611
[10]Fan N Y,Chen Y J,Feng Q M,et al.J.Mater.Res.,2012,27:1471-1475
[11]Yu H G,Zhu Z F,Zhou J H,et al.Appl.Surf.Sci.,2013,265:424-430
[12]Dai Z,Qin F,Zhao H P,et al.ACS Catal.,2016,6:3180-3192
[13]Shi R,Huang G L,Lin J,et al.J.Phys.Chem.C,2009,113:19633-19638
[14]Li J Q,Guo Z Y,Liu H,et al.J.Alloys Compd.,2013,581:40-45
[15]Yang J,Wang X H,Zhao X L,et al.J.Phys.Chem.C,2015,119,3068-3078
[16]Liu L,Ding L,Liu Y G,et al.Appl.Surf.Sci.,2016,364,505-515
[17]Cui W Q,An W J,Hu J S,et al.J.Hazard.Mater.,2014,280:417-427
[18]Cui W Q,An W J,liu L,et al.Mater.Lett.,2014,132:251-254
[19]Tian J,Hao P,Wei N,et al.ACS Catal.,2015,5:4530-4536
[20]Zhang F J,Zhu S F,Xie F Z,et al.Sep.Purif.Technol.,2013,113:1-8
[21]Huang J,Tan G Q,Ren H J,et al.ACS Appl.Mater.Interfaces,2014,6:21041-21050
[22]Huang H W,Liu K,Chen K,et al.J.Phys.Chem.C,2014,118:14379-14387
[23]Xu H,Xu Y G,Li H M,et al.Dalton Trans.,2012,41(12):3387-3394
[24]Zhang M Y,Shao C L,Mu J B,et al.CrystEngComm,2012,14(2):605-612
[25]Tian J,Sang Y H,Yu G W,et al.Adv.Mater.,2013,25:5075-5080
[26]Wang D J,Guo L,Zhen Y Z,et al.J.Mater.Chem.A,2014,2(30):11716-11727
[27]He Y,Zhang Y H,Huang H W,et al.Inorg.Chem.Commun.,2014,45(8):55-58
[28]Huang H W,Chen G,Zhang Y H.Inorg.Chem.Commun.,2014,44(45):46-49
[29]Hu X F,Mohamood T,Ma W H,et al.J.Phys.Chem.B,2006,110:26012-26018
[30]Xiang Q J,Yu J G,Jaroniec M.J.Phys.Chem.C,2011,115:7355-7363
[31]Hosseini Z,Taghavinia N,Sharifi N,et al.J.Phys.Chem.C,2008,112(47):18686-18689
[32]Bai X J,Wang L,Zong R L,et al.Langmuir,2013,29(9):3097-3105
[33]Ji P F,Zhang J L,Chen F,et al.Appl.Catal.,B,2009,85(3/4):148-154
[34]Yin M,Li Z,Kou J,et al.Environ.Sci.Technol.,2009,43(21):8361-8366
[35]Jiang H Y,Liu J J,Cheng K,et al.J.Phys.Chem.C,2013,117:20029-20036
[36]Wang D J,Shen H D,Li Guo,et al.RSC Adv.,2016,6:71052-71060
[37]DONG Wei-Xia(董偉霞),BAO Qi-Fu(包啟富),GU Xing-Yong(顧幸勇),et al.Chinese J.Inorg.Chem.(無機化學學報),2017,33(20):292-298
Construction of Bi2WO6Quantum Dots(QDs)Decorated Bi2MoO6-xF2xHeterostructures with Enhanced Photocatalytic Activity
WANG Dan-Jun*,1SHEN Hui-Dong1FU Meng-Xi1,2WANG Chan1GUO Li1YANG Xiao1FU Feng1
(1College of Chemistry&Chemical Engineering,Yan′an University,Shaanxi Key Laboratory of Chemical Reaction Engineering,Shaanxi,Yan′an 716000,China)
(2School of materials science and chemical engineering,Xi′an Technological University,Xi′an 710021,China)
Bi2WO6@Bi2MoO6-xF2xheterostructures have been successfully synthesized via a facile precipitationdeposition process.XRD,XPS,FE-SEM,HR-TEM,EDS,UV-Vis-DRS,PC and EIS techniques were employed to analyze the phase composition,morphology,light absorption and photoelectrochemical properties of as-synthesized samples.Photocatalytic degradation of model pollutant RhB was selected as a probe reaction to investigate the photocatalytic activities enhanced mechanism of Bi2WO6@Bi2MoO6-xF2xheterostructures.The morphology analysis indicated that the as-synthesized Bi2MoO6microsphere is composed of massive nanoplate with the thickness of 20~50 nm,and monodispersed Bi2WO6quantum dots(QDs)with an average size of 10 nm was deposited on the surface of Bi2MoO6microsphere.Comparison to pure Bi2MoO6and Bi2WO6,1∶1Bi2WO6@Bi2MoO6-xF2xheterostructures exhibits the best photocatalytic activity and photoelectrochemical property.The transient photocurrent responses and EIS spectra analysis results indicate that Bi2WO6QDs deposition can improve the separation sufficiency andmigration rate of photogeneraged electrons/holes.The radical scavengers test further confirm that·and h+are the main reactive species during the photocatalytic process.On the basis of experimental results,the mechanism of the enhanced photocatalytic activity for Bi2WO6@Bi2MoO6-xF2xheterostructure via synergistic effect of F-doping and Bi2WO6QDs deposition co-modification was also discussed.
precipitation-deposition process;quantum dots(QDs)decoration;Bi2WO6/Bi2MoO6-xF2xheterostructure;synergistic effect;photocatalysis;synergistic effect
TB333
A
1001-4861(2018)01-0073-10
10.11862/CJIC.2018.029
2017-08-04。收修改稿日期:2017-10-10。
國家自然科學基金(No.21666039,21663030),陜西省科技廳基金(No.2013SZS20-P01,2015SF291),陜西省教育廳基金項目(No.15JS119)和陜西省大學生創(chuàng)新創(chuàng)業(yè)訓練計劃項目(No.1517,1563)資助。
*通信聯(lián)系人。 E-mail:wangdj761118@163.com
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
