W20O58(010)表面氫吸附機理的第一性原理研究?
2018-01-18 19:01:12姜平國汪正兵閆永播劉文杰
物理學報 2017年24期
關鍵詞:結構
姜平國 汪正兵 閆永播 劉文杰
(江西理工大學冶金與化學工程學院,贛州 341000)
(2017年6月17日收到;2017年9月9日收到修改稿)
1 引 言
W20O58是當前制取超細鎢粉最為廣泛的原材料[1?7].鎢具有優異的物理和化學特性,如高熔點、高硬度、高密度、高彈性模量、優良的斷裂韌性及良好的導熱性等性質,是生產多種重要功能和構型材料的主要原料,從而被廣泛用于硬質合金、鎢基高密度合金、鎢絲、鎢電極等眾多領域[8?10].隨著現代工業技術的發展,傳統鎢粉末制備而成的鎢制品已難以滿足工業需要,然而采用超細鎢粉制取的鎢制品提高了其強度、韌性,降低了金屬塑性-脆性轉變溫度,大大改善了材料的性能[11?13].
近年來,許多研究者對氫氣還原藍鎢制取超細鎢粉的熱力學和還原工藝進行了大量研究[7,14,15],藍鎢是目前制取鎢粉使用最廣泛的原料,吳曉東等[7]以熱分解新型鎢酸鹽得到的純組分藍鎢(四方晶型)為原料,在低溫、干燥、高氣流量、薄料層的條件下,制備了性能優于黃鎢制備的超細鎢粉.然而對其微觀擴散吸附動力學方面的研究未曾有報道,因此有必要對其進行理論研究.
事實上,密度泛函理論是計算物理和化學的重要工具之一,它已經成功地應用于固體功能材料的結構和性質[16?19],氣體及有機化合物在固體表面的吸附性質[20,21]以及表面微觀反應機理[22]的研究.本文采用基于密度泛函理論的第一性原理方法,對晶胞W20O58,W20O58(010)表面結構及表面氫吸附機理進行了理論計算,以期為深入認識W20O58晶體特性、W20O58(010)表面特性及與H2分子反應規律提供有益的理論支持.
2 計算模型及方法
2.1 W20O58結構模型
W20O58晶體結構模型如圖1所示,它是單斜的不規則非化學計量比結構,具有非規則對稱結構,其空間群是P2/m[23],由W和O構成的八面體,W和O分別處于八面體的中心位置和頂點位置,其中每個W原子含有6個配位,O原子含有2個配位.
2.2 W20O58(010)結構模型
前人研究表明W20O58的(010)面表面能最低[24],于是本文創建了兩種不同表面的W20O58(010)表面結構,如圖2所示.圖2(a)為WO終止(010)表面結構,圖2(b)為O終止(010)表面結構;圖中W5c和W6c分別為5配位和6配位的W原子,O1c和O2c分別為1配位和2配位的O原子.每個表面結構都含有5層原子,原子層之間設置了厚度為1.0 nm的真空層,真空層厚度和原子層均通過了收斂性測試.
根據W20O58與H2反應的熱力學[7],最終得到W和H2O,其中WO終止(010)表面有O1c和O2c兩種配位的O原子,O終止(010)表面只有O1c一種配位的O原子,于是創建了如圖3所示的6種不同吸附方式的H2分子吸附構型,其中圖3(a)和圖3(b)分別為WO終止(010)表面結構H2分子水平(level)和垂直(vertical)吸附于O1c位上,分別記為WO-L-O1c和WO-V-O1c;圖3(c)和圖3(d)分別為WO終止(010)表面結構H2分子水平和垂直吸附于O2c位上,分別記為WO-L-O2c和WO-VO2c;圖3(e)和圖3(f)分別為O終止(010)表面結構H2分子水平和垂直吸附于O1c位上,分別記為O-L-O1c和O-V-O1c.
2.3 計算方法
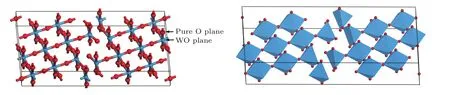
圖1 (網刊彩色)W20O58晶體結構示意圖(紅球表示O,藍球表示W)Fig.1.(color online)Schematic diagram of W20O58crystal structure.Red and blue balls denote O and W atoms,respectively.
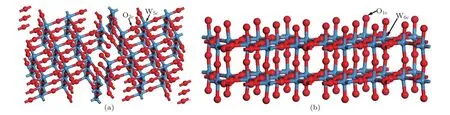
圖2 (網刊彩色)W20O58(010)表面結構(紅球表示O,藍球表示W) (a)WO終止(010)表面結構;(b)O終止(010)表面結構Fig.2.(color online)W20O58(010)surface structure(red and blue balls denote O and W atoms,respectively):(a)WO-terminated(010)surface structure;(b)O-terminated(010)surface structure.
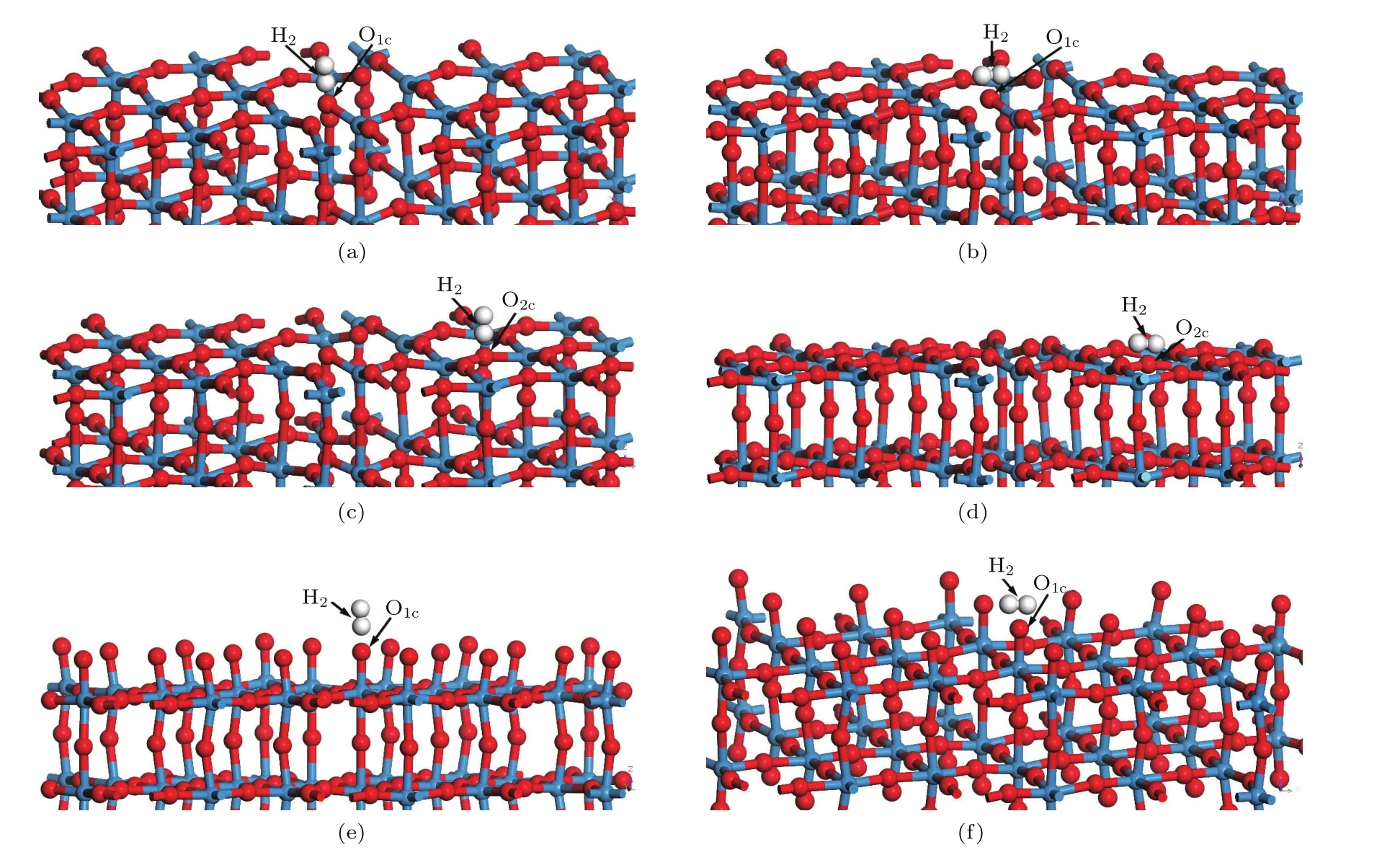
圖3 (網刊彩色)W20O58(010)表面吸附H2分子構型(紅球表示O,藍球表示W) (a)WO終止(010)表面結構,WOL-O1c;(b)WO終止(010)表面結構,WO-V-O1c;(c)WO終止(010)表面結構,WO-L-O2c;(d)WO終止(010)表面結構,WO-V-O2c;(e)O終止(010)表面結構,O-L-O1c;(f)O終止(010)表面結構,O-V-O1cFig.3.(color online)H2molecule adsorption structures on W20O58(010)surface(red and blue balls denote O and W atoms,respectively):(a)WO-terminated(010)surface structure,WO-L-O1c;(b)WO-terminated(010)surface structure,WO-V-O1c;(c)WO-terminated(010)surface structure,WO-L-O2c;(d)WO-terminated(010)surface structure,WO-V-O2c;(e)O-terminated(010)surface structure,O-L-O1c;(f)O-terminated(010)surface structure,O-V-O1c.
所有計算工作都是由軟件Materials Studio 7.0中的CASTEP(Cambridge serial total energy package)[25]模塊完成,采用廣義梯度近似中的PW91(Perdew-Wang1991)方法來處理交換關聯能[26,27].采用Monkhorst-Pack[28]方法來設置k點網格數目,運用Vanderbilt超軟贗勢來處理電子之間的相互作用力.對平面波截斷能和k點網格數進行了收斂性測試,對W20O58晶胞、W20O58(010)表面結構和其表面吸附H2分子結構的平面波截斷能均設置為350 eV,k點網格數均設置為2×5×1.幾何構型優化采用了Broyden-Fletcher-Goldfarb-Shanno算法[29],其收斂判別準則和能量計算精度均采用 fi ne,即費米(smearing)值為0.1 eV,體系總能量收斂判據為1.0×10?5eV/atom,每個原子上的力收斂判據為0.3 eV/nm,位移收斂判據為0.0001 nm,應力偏差小于0.05 GPa,允許所有原子弛豫.所有計算均在倒易空間中和絕對零度下進行.色散校正和零點能對吸附能的影響較小,忽略二者影響.W的原子軌道為5s25p65d46s2,O的原子軌道為2s22p4,H的原子軌道為1s.
3 計算結果與討論
3.1 W20O58晶體結構性質
將W20O58晶體結構優化并進行能量計算,其晶格常數為a=1.2181 nm,b=0.3866 nm,c=2.3839 nm,β=94.39°,與實驗值[25]基本一致,且得到如圖4所示的沿著布里淵區高對稱點方向的能帶結構(band structure)[30]和圖5的態密度(density of states,DOS),能帶結構圖和DOS圖中將能量零點取為費米面,作為能量的參考點.
從圖4所示的能帶結構可以得到導帶底為?0.49 eV,價帶頂為?1.29 eV,帶隙為0.8 eV,是間接帶隙.軟件默認將能帶結構中的費米能級設置在零點.從圖4可以看出費米能級在帶隙之上且穿過部分能帶,則表示W20O58晶體具有金屬性,而實驗中測出其具有半導體性可能是由于表面接觸了氧原子.WOx中x越大電荷載流子濃度越小,費米能級向帶隙移動,由金屬性轉變為半導體性.由圖5的總態密度(total density of states,TDOS)和分態密度(partial density of states,PDOS)可以看出W18O49導帶主要是來自O-2p和W-5d電子貢獻.W18O49價帶主要是由三部分組成:?8.6—?1.3 eV的價帶主要由O-2p和部分W-5d的電子貢獻;?21—?17 eV范圍內的價帶,這一部分主要由O-2s態和少量W-5d態形成;價帶?40—?38 eV和?77—?75 eV處分別存在一部分孤立能帶,這兩處孤立能帶分別由W-5p和W-6s態形成.
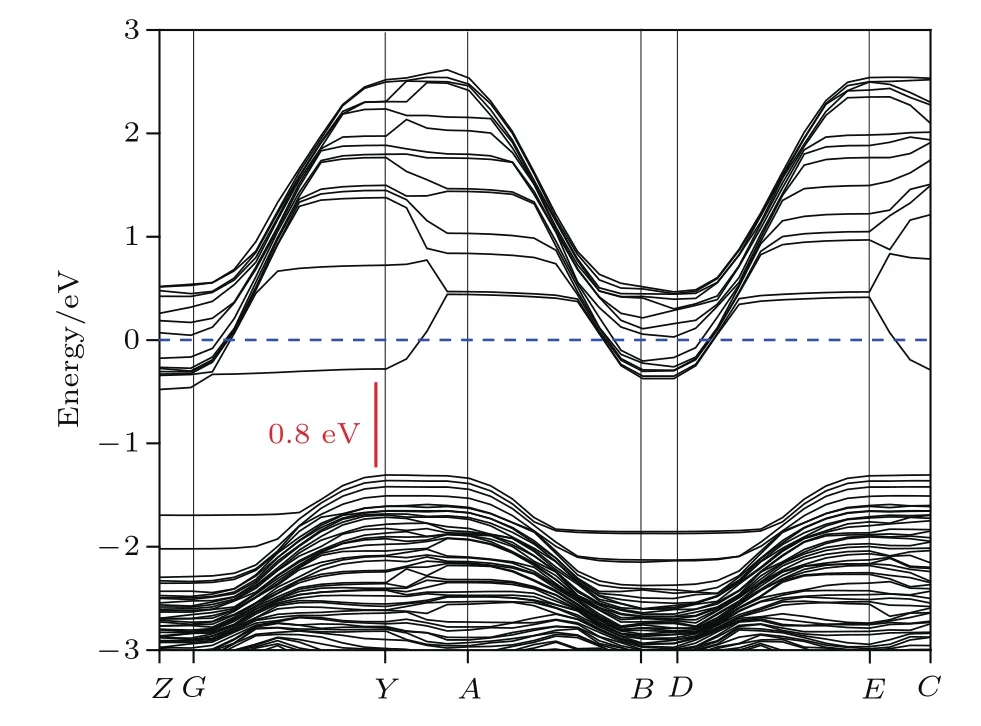
圖4 W20O58晶體能帶結構Fig.4.Band structure of W20O58crystal.
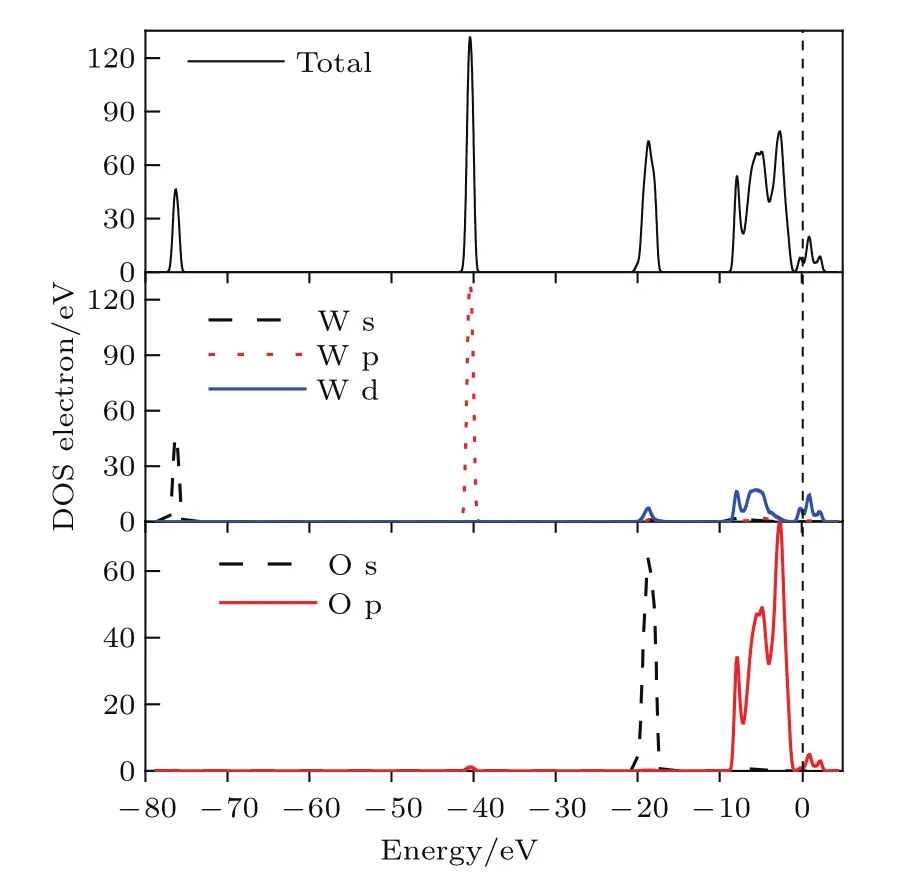
圖5 (網刊彩色)W20O58晶體TDOS和PDOSFig.5.(color online)TDOS and PDOS of W20O58crystal.
從能帶圖4中可以看出W20O58能帶的導帶底和價帶頂較寬,說明成鍵較強;能帶不平緩,能帶較寬表示電子較活躍.費米能級EF進入導帶,費米能級EF處的DOS不為零,是有具體數值的,則費米面與能帶相交叉,產生了部分填充的能帶,具有導電性的金屬特性.費米能級EF兩側總共有三個尖峰,尖峰之間的DOS不為零,為贗能隙.從DOS圖5中可以看出在?8.6—?1.3 eV和?1.3—?2.7 eV處的帶寬較大,與能帶結構圖相符合,表明電子離域性較強,成鍵強,組成這條能帶的原子軌道擴展性比較強;從圖5中W原子和O原子的PDOS,可以看到二者并不是孤立的,電子DOS重疊較多,說明在W20O58體系中W—O相互作用較強,以共價鍵居多.
3.2 W20O58(010)表面原子結構和電子結構
圖6為WO終止(010)表面和O終止(010)表面部分原子結構優化結果.從圖6可以看出WO終止(010)表面的部分原子結構的第一層和第三層的O2c原子向外凸出,O終止(010)表面部分原子結構O2c原子有向內凹陷也有向外凸出,這是由于O原子之間庫侖排斥作用引起的.從WO終止(010)表面部分原子結構圖可以看出,表面的O2c原子相對于W原子向外凸出,于是形成如圖6(a)所示的鋸齒狀的W—O—W鏈型結構,W—O—W鍵角分別為153.216°和152.183°.從O終止(010)表面部分原子結構圖可以看出,表面的O1c原子向外側移動,使得與其相連的W原子也向外側移動,形成長短相間的W—O鍵,同樣形成如圖6(b)所示的鋸齒狀的W—O—W鏈型結構,W—O—W鍵角分別為171.357°和170.774°.庫侖力的作用使得WO終止(010)表面和O終止(010)表面鋸齒狀的W—O—W鏈型結構處的W—O鍵長相比較優化前鍵長都會改變.這些現象表明 W—O鍵長和W—O—W鍵角的改變是表面原子弛豫的主要方式.
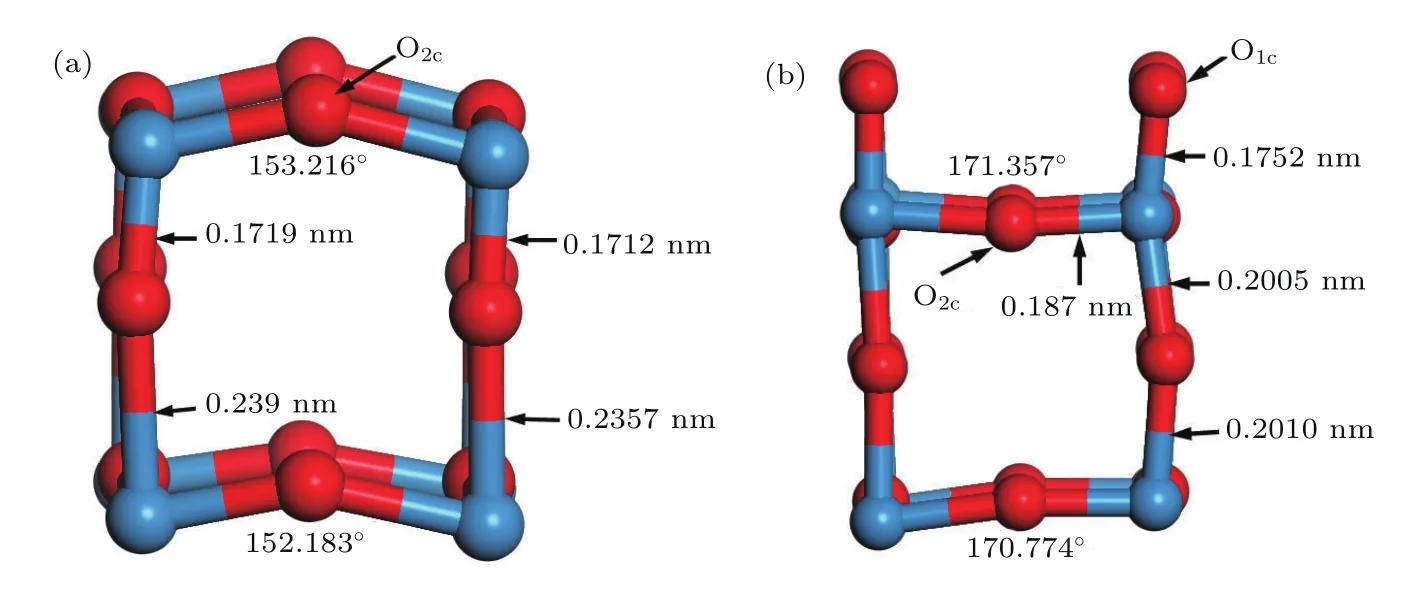
圖6 (網刊彩色)W20O58(010)表面部分弛豫原子結構(紅球表示O,藍球表示W) (a)WO終止(010)表面;(b)O終止(010)表面Fig.6.(color online)Part of geometrical structure of W20O58(010)surface(red and blue balls denote O and W atoms,respectively):(a)WO-terminated(010)surface;(b)O-terminated(010)O surface.
圖7所示為WO終止(010)表面結構和O終止(010)表面結構優化后DOS圖.從圖7可見,因為O經過雜化后,s軌道和p軌道都缺少一個電子,W是sp3d2雜化.WO終止(010)表面結構的導帶主要是由W-5d和O-2p電子雜化,費米能級處于導帶內,導帶內有部分電子填充,且附近主要由W-5d電子貢獻;其價帶在?9—?1 eV處主要是由W-5d上的單電子和O-2p單電子相互作用雜化,在此能量區間內有4個成鍵峰,O-2p和W-5d電子軌道雜化作用較強,相互作用明顯;在WO終止面,在?21.5—?17 eV處是由O-2s和少量的W-5d電子相互作用,此能量區間內有3個成鍵峰,少部分的O-2s和W-5d電子軌道重疊,發生電子轉移,組成化學鍵.O終止(001)表面結構的導帶主要是由W-5d和O-2p電子貢獻,雜化了微量的W-5p電子態,費米能級處于價帶內,存在空穴,且附近主要由O-2p電子貢獻;其價帶在?7—0.8 eV處是主要由O-2p和W-5d電子相互作用,有5個成鍵峰,相互作用強,共價鍵較多;在?18.6—?14.7 eV處主要是由O-2s貢獻,且雜化了少量的W-5d電子與微量的W-5p和W-6s電子,有2個成鍵峰,相互作用較弱.
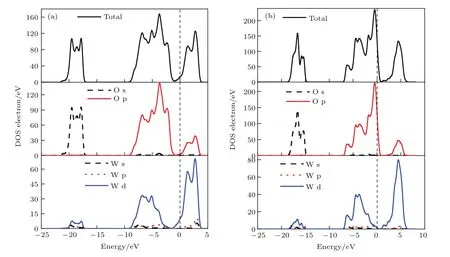
圖7 (網刊彩色)W20O58(010)表面結構的TDOS和PDOS (a)WO終止(010)表面結構;(b)O終止(010)表面結構Fig.7.(color online)TDOS and PDOS of W20O58(010)surface structures:(a)WO-terminated(010)surface structure;(b)O-terminated(010)surface structure.
3.3 H2在W20O58(010)表面的吸附與解離
在一定條件下,H2分子首先會解離成H原子,然后H原子會吸附在W20O58表面,最后會發生化學反應,解離吸附是產生化學反應的前提.為比較H2在不同表面不同吸附位置的吸附性能,探討了如圖3所示的六個吸附位置.
為了探究不同吸附構型的穩定性,首先計算各個吸附構型的吸附能,吸附能Eads的表達式為[31]
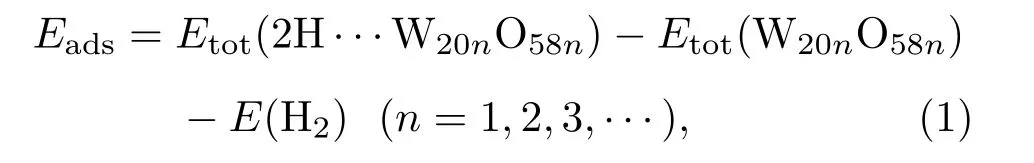
其中Etot(2H···W20nO58n)是W20O58(010)表面吸附2個H原子后的總能,Etot(W20nO58n)是純凈W20O58(010)表面結構的總能,E(H2)是將一個氫分子置于晶格常數為1 nm的立方晶格中計算所得到的能量,其中設置平面波截斷能為400 eV,k點網格數為8×8×8,收斂精度為 fi ne,spin設置為2.0,初始自旋值為1mu_B.將H2分子放入1 nm的立方晶格中,此原胞足夠大,近鄰原胞影響可忽略不計.計算得到H2分子的H—H鍵長為0.0754 nm,與實驗值0.0751 nm相差較小;單個氫分子的總能為?6.67 eV.同樣的方式算得單個氫原子的總能為?1.09 eV.用結合能公式Ebinding=?(EH2?2EH),得到H2分子的結合能Ebinding為4.58 eV,與實驗值4.75 eV相差不大.因此,可以認為這樣的模擬是準確可信的.
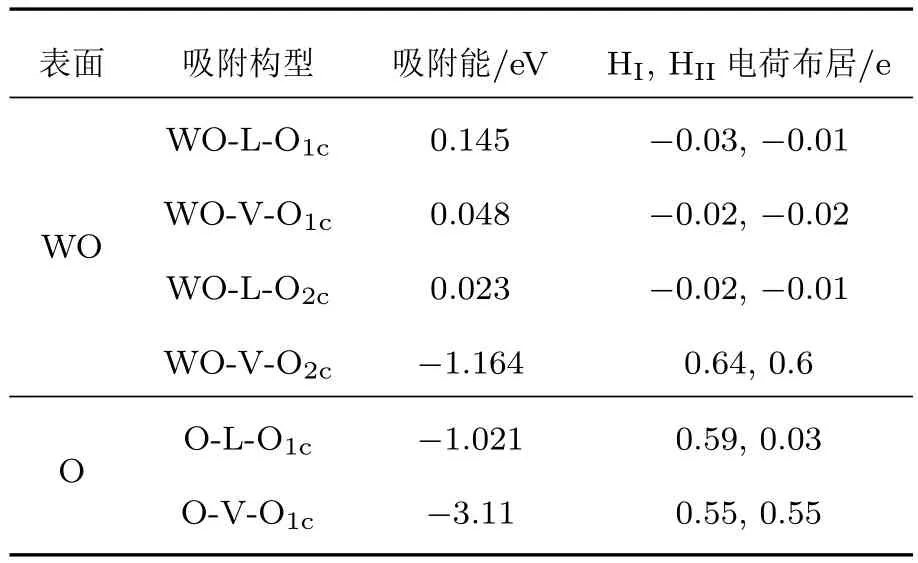
表1 W20O58(010)表面吸附氫分子后的結構參數變化Table 1.Variance of structural parameters after H2 adsorption on W20O58(010)surface.
表1所列為四種吸附構型的變化參數,其中吸附能是由上述吸附能計算(1)式得到.表2為W20O58(010)表面被吸附的H2分子中兩個H原子間的鍵長DH—H,兩個H原子與吸附位M間的間距DM-HI和DM-HII參數.由表1和表2可以看出WO終止(010)表面的WO-L-O1c,WO-V-O1c,WO-LO2c三種吸附構型的吸附能均為正值,H—H鍵的鍵長分別為0.0754,0.0751,0.0753 nm,與初始的0.0754 nm相比變化非常小,氫氣分子并未發生解離,且氫氣分子中的2個H原子與吸附位間的間距相比于吸附前間距越來越大,說明氫氣分子不會解離吸附在表面,而是漸漸遠離吸附構型表面.因此吸附能、氫鍵的變化以及吸附間距變化三個方面表明這三種吸附構型的吸附效應都是非常微弱的且不穩定.對于WO-V-O2c,O-L-O1c,O-V-O1c這三種吸附構型都很穩定,吸附能均為負值,分別為?1.164,?1.021,?3.11 eV,H—H鍵的鍵長分別為0.1614,0.6883,0.1564 nm,與初始的H—H鍵鍵長相比變化非常大,H—H鍵斷裂;通過H原子電荷布居分析,WO-V-O2c吸附構型的兩個氫原子分別向吸附位O原子和W原子提供了0.64e,0.6e電子,O-L-O1c吸附構型的兩個氫原子只有一個H原子向吸附位O原子提供了0.59e電子,而O-V-O1c吸附構型的兩個氫原子都向吸附位O原子提供了0.55e電子;這三種穩定的吸附構型中最穩定的吸附構型是O-V-O1c,氫氣分子解離后,兩個氫原子吸附到同一個O1c原子上,形成H—O鍵,由于處于O終止(010)表面的最外層的O1c原子含一個不飽和化學鍵,因此易于H原子結合形成H—O鍵.
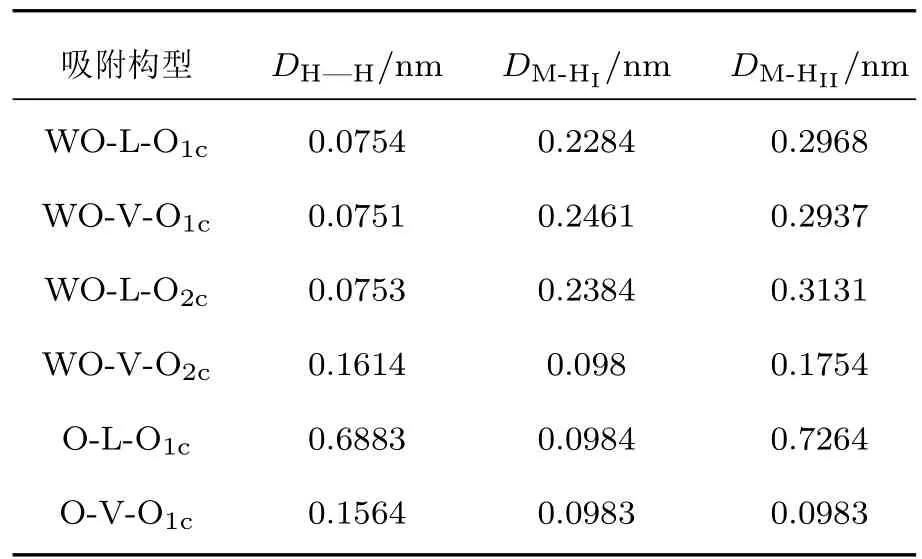
表2 W20O58(010)表面被吸附的H2分子中兩個H原子間的鍵長DH—H,兩個H原子與吸附位M間的間距DM-HI和DM-HIITable 2.Bond lengths of H—H bond(DH—H),distances between two H atoms of the adsorbed H2and the adsorption sites(DM-HIand DM-HII)for H2adsorption on W20O58(010)surface.
圖8為WO-V-O2c,O-L-O1c,O-V-O1c三種穩定吸附構型氫氣分子吸附前后側面圖.從圖8可以看出,在這三種吸附構型中無論H2分子水平吸附,還是垂直吸附于吸附位上后,H—H鍵斷裂,H2分子解離,形成兩個H原子;對于WO-V-O2c吸附構型,氫氣分子解離后的兩個H原子HI,HII分別與吸附位O2c和W原子成鍵,形成H—O鍵和W—H鍵,且吸附位O2c和W原子的鍵斷裂,W4c和O1c原子成鍵,附近部分W原子和O原子有成鍵,也有斷鍵.對于O-L-O1c吸附構型,氫氣分子解離后的H原子HI與吸附位O1c形成H—O鍵,而H原子HII遠離了O終止(010)表面,W6c原子與吸附位O1c相連接的W—O鍵鍵長從0.1737 nm延長到了0.191 nm,增幅不大,W—O鍵沒有斷裂,吸附位O1c原子吸附一個HI原子后轉變為O2c原子.對于O-V-O1c吸附構型,氫氣分子解離后的兩個H原子都與吸附位O1c原子形成H—O化學鍵,W6c原子與吸附位O1c相連接的W—O鍵長從0.1737 nm增大為0.2445 nm,增幅較大,W—O鍵斷裂,從而在表面頂部形成一個水分子,所得到H—O鍵的鍵長(0.0983,0.0983 nm)和H—O—H鍵的鍵角(105.48°)均與水分子在基態時的鍵長(0.0957—0.1 nm)與鍵角(104.52°—109.5°)基本一致;W—O鍵斷裂也使得W6c轉變為W5c,致使表面形成了一個氧空位.
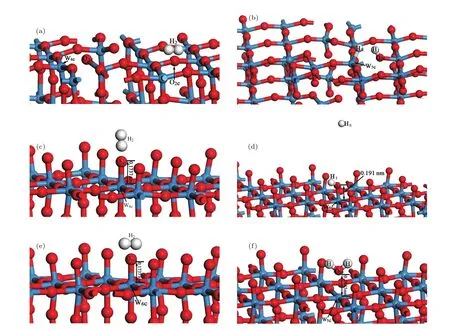
圖8 (網刊彩色)三種穩定吸附構型H2分子吸附前后的結構(紅球表示O,藍球表示W) (a),(b)WO-V-O2c吸附構型;(c),(d)O-L-O1c吸附構型;(e),(f)O-V-O1c吸附構型Fig.8.(color online)The con fi gurations before and after H2adsorption on stable adsorption con fi guration(red and blue balls denote O and W atoms,respectively):(a),(b)WO-V-O2cadsorption con fi guration;(c),(d)O-L-O1cadsorption con fi guration;(e),(f)O-V-O1cadsorption con fi guration.
3.4 穩定吸附構型H2分子吸附的DOS分布
H2分子吸附在W20O58(010)表面不僅影響其表面結構,同時也會改變其電子結構.通過對比H2分子在WO-V-O2c,O-L-O1c和O-V-O1c吸附構型吸附前后的DOS分析了H2分子其表面吸附原子之間相互作用的情況.
圖9(a)和圖9(b)由上到下分別給出了WOV-O2c吸附構型氫氣分子吸附前后的TDOS,O原子與W原子PDOS,H2分子的DOS.對比兩圖可以看出WO-V-O2c構型吸附前后的DOS發生了變化,特別是H2分子水平吸附到WO終止(010)表面后,DOS發生了很大的變化,H原子的1s軌道峰向左移動,降低了H的1s軌道的能量,在?21.3—?19 eV處出現了兩個成鍵峰,這是HI的1s軌道與O原子的2s發生軌道重疊;在?9 eV處出現了一個大的尖峰,這是由于HI的1s軌道與O原子的2p發生雜化作用,而在?2.6 eV處出現的大尖峰是由HII的1s軌道與W原子的5d軌道雜化作用造成的;在?8.4—?3.5 eV和0—3.6 eV處產生的成鍵峰,是由HI的1s軌道與O的2p發生sp軌道雜化作用和HII的1s軌道與W的5d軌道雜化作用共同造成的.因此氫氣分子解離后,兩個H原子HI,HII分別與O和W原子形成共價鍵.

圖9 (網刊彩色)WO-V-O2c吸附構型H2分子吸附前后的TDOS和PDOS (a)吸附前;(b)吸附后Fig.9.(color online)TDOS and PDOS(a)before and(b)after H2adsorption on WO-V-O2c.
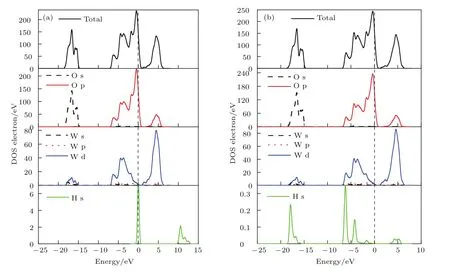
圖10 (網刊彩色)O-L-O1c吸附構型H2分子吸附前后的TDOS和PDOS (a)吸附前;(b)吸附后Fig.10.(color online)TDOS and PDOS(a)before and(b)after H2adsorption on O-L-O1c.
圖10(a)和圖10(b)由上到下分別給出了O-LO1c吸附構型氫氣分子吸附前后的TDOS,O原子與W原子PDOS,H2分子的DOS.對比兩圖可以看出O-L-O1c構型吸附前后TDOS能量值基本不變,但氫氣分子水平吸附到O終止(010)表面后,解離形成兩個氫原子HI,HII,其中HI的1s軌道峰向左移動,能量降低,在?18,?16.7 eV處H的1s軌道與O的2s軌道發生軌道重疊,分別形成了一個較強和一個較弱的成鍵峰;在?6.1,?4.2 eV處HI的1s軌道與O的2p軌道發生較強sp軌道雜化作用,形成了兩個較強的成鍵峰,在?1.7,4.2,5.3 eV處HI的1s軌道與O的2p軌道發生較弱軌道雜化作用,形成了三個較弱的成鍵峰.而HII原子的電子軌道并未發生變化.因此HI原子與O1c原子形成H—O鍵,HII原子遠離表面,沒有成鍵.
圖11(a)和圖11(b)由上到下分別給出了OV-O1c吸附構型氫氣分子吸附前后的TDOS,O原子與W原子PDOS,H2分子的DOS.對比兩圖可見氫氣的DOS發生了很大變化,H的1s軌道峰同樣向左移動,能量降低,在?19.5 eV處H的1s軌道與O的2s軌道重疊,產生了一個較強的孤立成鍵峰;H的1s軌道與O的2p軌道產生的sp軌道雜化作用使得在?7.1和6 eV處出現了兩個較強的成鍵峰,在?5.6和?4.4 eV處出現了兩個較弱的成鍵峰.因此氫氣分子解離后形成的兩個H原子都與吸附位O1c原子形成H—O化學鍵.
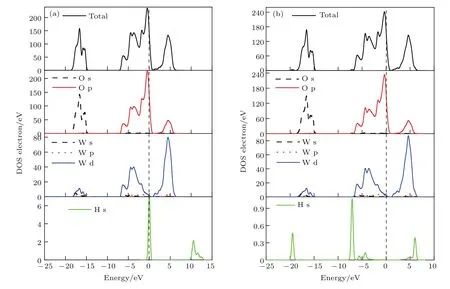
圖11 (網刊彩色)O-V-O1c吸附構型H2分子吸附前后的TDOS和PDOS (a)吸附前;(b)吸附后Fig.11.(color online)TDOS and PDOS(a)before and(b)after H2adsorption on O-V-O1c.
基于以上分析,最穩定的吸附構型是O-V-O1c,H2分子垂直吸附于O終止(010)表面后,會解離成兩個H原子吸附在同一個O1c位上,且H與O吸附作用較強,形成H—O化學鍵,這是由于O終止(010)表面O1c存在不飽和鍵,易與H結合形成共價的H—O鍵,致使2個H原子與O1c結合形成一個水分結構,而與O1c相連接的W6c原子轉變為W5c原子.此表面反應可以用以下化學方程式表示:
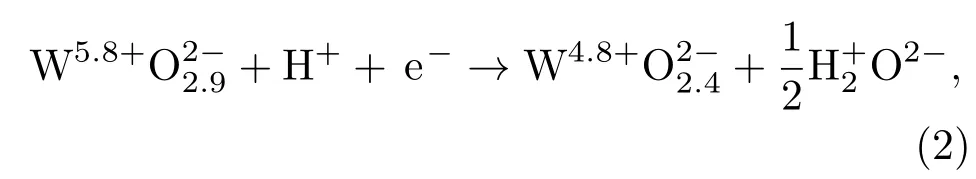
其中,此反應導致了W20O58表面氧的缺失并改變了其電子性質,從而造成其導帶被部分填充.
4 結 論
采用基于密度泛函理論的第一性原理方法研究了晶胞W20O58,W20O58(010)清潔表面與H2分子吸附在W20O58(010)表面的幾何結構和電子結構.研究發現W20O58(010)表面有WO終止(010)表面和O終止(010)表面兩種表面結構,結構優化后表面均形成長短相間的W—O鍵和鋸齒狀的W—O—W鏈型結構.H2分子吸附在W20O58(010)表面最穩定的吸附構型是O-V-O1c,即H2分子垂直吸附于O終止(010)表面后,會解離成兩個H原子吸附在同一個O1c位上,H的1s軌道與O-2p,2s發生相互作用,H與O形成化學鍵,生成了一個H2O分子結構,表面產生了一個氧空位.
[1]Peng W Z 2004Cemented Carbide21 142(in Chinese)[彭衛珍2004硬質合金21 142]
[2]Fu X M 2004Shanghai Nonferrous Met.25 149(in Chinese)[傅小明2004上海有色金屬25 149]
[3]Wu X D,Chai Y X,Fu X M,Fu M X,Dai Q X 2005Chinese J.Rare Met.29 570(in Chinese)[吳曉東,柴永新,傅小明,傅明喜,戴起勛2005稀有金屬29 570]
[4]Wang G,Li H H,Huang Z W,Chen M H,Wang Q K,Zhong W L 2009Rare Metal.Mat.Eng.38 548(in Chinese)[王崗,李海華,黃忠偉,陳美華,王慶康,鐘偉良 2009稀有金屬材料與工程38 548]
[5]Liao J Q,Huang Z F,Lü H B,Chen S Y,Zou Z Q 2000J.Cent.South Univ.Technol.31 51(in Chinese)[廖寄喬,黃志鋒,呂海波,陳紹衣,鄒志強 2000中南工業大學學報31 51]
[6]Wang Z X,Shu D X,Tang X H 1993Eng.Chem.Metall.14 224(in Chinese)[王志雄,舒代萱,唐新和 1993化工冶金14 224]
[7]Wu X D,Chai Y X,Fu M X 2005Cemented Carbide22 65(in Chinese)[吳曉東,柴永新,傅明喜 2005硬質合金22 65]
[8]Li H K,Yang J G,Li K 2010Tungsten Metallurgy(Changsha:Central South University Press)pp36–39(in Chinese)[李洪桂,羊建高,李昆 2010鎢冶金學 (長沙:中南大學出版社)第36—39頁]
[9]Yu G,Han Q G,Li M Z,Jia X P,Ma H A,Li Y F 2012Acta Phys.Sin.61 040702(in Chinese)[于歌,韓奇鋼,李明哲,賈曉鵬,馬紅安,李月芬2012物理學報61 040702]
[10]Qiu K Q,Wang A M,Zhang H F,Qiao D C,Ding B Z,Hu Z Q 2002Acta Metall.Sin.38 1091(in Chinese)[邱克強,王愛民,張海峰,喬東春,丁炳哲,胡壯麒 2002金屬學報38 1091]
[11]Hua J S,Jing F Q,Dong Y B,Tan H,Shen Z Y,Zhou X M,Hu S L 2003Acta Phys.Sin.52 2005(in Chinese)[華勁松,經福謙,董玉斌,譚華,沈中毅,周顯明,胡紹樓2003物理學報52 2005]
[12]Tan J,Zhou Z J,Zhu X P,Guo S Q,Qu D D,Lei M K,Ge C C 2012Trans.Nonferrous Met.Soc.China22 1081
[13]Liu H M,Fan J L,Tian J M,You F 2009China Tungsten Ind.24 29(in Chinese)[劉輝明,范景蓮,田家敏,游峰2009中國鎢業24 29]
[14]Hessel S,Shpigler Β,Botstein O 1993Rev.Chem.Eng.9 345
[15]Wu X W,Luo J S,Lu B Z,Xie C H,Pi Z M,Hu M Z,Xu T,Wu G G,Yu Z M,Yi D Q 2009Trans.Nonferrous Met.Soc.China19 785
[16]Xu L,Yan Q Z,Xia M,Zhu L X 2013Int.J.Refract.Met.Hard Mater.36 238
[17]Yu Y X 2013Phys.Chem.Chem.Phys.15 16819
[18]Yu Y X 2016J.Phys.Chem.C120 5288
[19]Yang G M,Xu Q,Li B,Zhang H Z,He X G 2015Acta Phys.Sin.64 127301(in Chinese)[楊光敏,徐強,李冰,張漢壯,賀小光2015物理學報64 127301]
[20]Xue L,Ren Y M 2016Acta Phys.Sin.65 156301(in Chinese)[薛麗,任一鳴 2016物理學報 65 156301]
[21]Yu Y X 2014ACS Appl.Mater.Interfaces6 16267
[22]Li B,Wu T Q,Wang C C,Jiang Y 2016Acta Phys.Sin.65 216301(in Chinese)[李白,吳太權,汪辰超,江影2016物理學報65 216301]
[23]Chen J,Lu D Y,Zhang W H,Xie F Y,Zhou J,Gong L,Liu X,Deng S Z,Xu N S 2008J.Phys.D:Appl.Phys.41 115305
[24]Lu D Y,Chen J,Deng S Z,Xu N S,Zhang W H 2008J.Mater.Res.23 402
[25]Segall M D,Lindan P J D,Probert M J,Pickard C J,Hasnip P J,Clark S J,Payne M C 2002J.Phys.:Condens.Matter14 2717
[26]Wang Y,Perdew J P,Chevary J A,Macdonald L D,Vosko S H 1990Phys.Rev.A41 78
[27]Perdew J P,Chevary J A,Vosko S H,Jackson K A,Pederson M R,Singh D J 1992Phys.Rev.B46 6671
[28]Monkhorst H J,Pack J D 1976Phys.Rev.B13 5188
[29]Fletcher R 1970Comput.J.13 317
[30]Setyawan W,Curtarolo S 2010Comput.Mater.Sci.49 299
[31]Yamaguchi O,Tomihisa D,Kawabata H,Shimizu K 1987J.Am.Ceram.Soc.70 94
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
