基于多重進化矩陣的蛋白質特征向量構造方法①
2018-03-02 06:16:14杜月寒鹿文鵬劉毅慧成金勇
計算機系統應用 2018年2期
關鍵詞:結構
杜月寒,鹿文鵬,劉毅慧,成金勇
(齊魯工業大學(山東省科學院)信息學院,濟南 250353)
蛋白質是生物體內生命活動的主要承擔者,是一切生命活動的基礎,它的生理功能除了體現在氨基酸構成上還體現在它的空間結構上[1].因此,預測蛋白質結構是生物信息學領域的一個重要任務.通常,蛋白質結構包括4個層次[2]:一級結構即氨基酸的排列順序;二級結構主要是由氫鍵維持的α-螺旋和β-折疊;三級結構是完全折疊的蛋白質的空間結構殘基的立體排列模式;四級結構是多個蛋白質亞基組成的蛋白質復合體的結構(即蛋白質之間的交互作用).蛋白質二級結構是聯系蛋白質一級結構和三級結構的紐帶,而且也是從一級結構預測其三級結構的關鍵步驟[3,4].當蛋白質二級結構預測正確率達到80%時,就可以準確預測一個蛋白質分子的三維空間結構[5].可見,蛋白質二級結構預測已經成為研究蛋白質結構和功能的重要手段.
由于已測定結構的蛋白質數量遠遠小于已知的蛋白質序列數量[6],并且傳統的生物實驗測定蛋白質結構的方法花費昂貴且耗時時間較長.因此,采用數據驅動的方法(如機器學習技術)來預測未知的蛋白質的結構和功能廣受青睞.在過去的一段時間內,很多方法被提出來用于蛋白質結構類的預測.而影響蛋白質結構類預測效果的關鍵因素主要集中在兩個方面上:一是分類預測算法,Zhou等人使用神經網絡[7],Mandle等人使用支持向量機[8],Wang和Peng等使用深度卷積神經網絡技術來進行蛋白質結構預測[9];二是蛋白質特征信息提取,如Chou等人提出的偽氨基酸組成(PseAA法)[10-12],Cao等人提出的基于簡化PSSM與蛋白質結構位置信息的特征表示算法[13].
一般的預測方法通常使用BLOSUM62矩陣構造特征向量,對蛋白質進化過程中存在的氨基酸位點突變現象缺乏考慮.本文提出一種新的特征表示方法,對于一條蛋白質序列,同時使用多種進化趨異度的矩陣來表示蛋白質序列,更全面的考慮了殘基替換的可能性.不同的進化矩陣對不同相關程度的蛋白質序列的敏感性不同.這使得多重進化矩陣這種蛋白質序列特征表示方法,不僅可以很好地反映序列中氨基酸的位置信息,而且全面考慮序列內部近相關和遠相關蛋白質區域[14]之間的相互影響.本文結合交叉驗證法和網格搜索法來確定實驗參數,先在大范圍大步距粗搜,初步確定一個最優參數區間,之后在此區間進行小步距精搜,結合網格搜索法和交叉驗證法共同確定實驗參數.在數據集RS126、CB513和25PDB上進行的多組實驗,表明本文所提出的基于多重進化矩陣的特征向量構造方法能夠有效提高蛋白質二級結構預測精度.
1 相關知識
1.1 位置特異性矩陣
通過蛋白質序列的位置特異性打分矩陣而不是僅依靠序列來預測蛋白質結構,是公認的提高預測精度的方法.序列的多重比對反映了蛋白質家族的共同特征,提取了結構的保守信息及家族中特定的殘基替換模式,同時多序列比對所攜帶的進化信息也表明了蛋白質進化過程中的相互作用[15].
本文主要采用了BLAST的本地化使用來獲得蛋白質序列的profile.將BLAST軟件包下載到本地以后,可以通過命令行的形式去調用相應的可執行文件.在這里,我們使用PSI-BLAST程序(h=0.001,j=3)搜索和調整無冗余的NR數據庫.該程序將返回一個20維矢量的PSSM[16],其值是20個氨基酸保守的突變分數.這樣得到的PSI-BLAST profile是一個L×20的矩陣(其中L是蛋白質序列的長度),也稱之為位置特異性打分矩陣(Position-Specific Score Matrix,PSSM).PSSM矩陣形式如公式(1)所示.
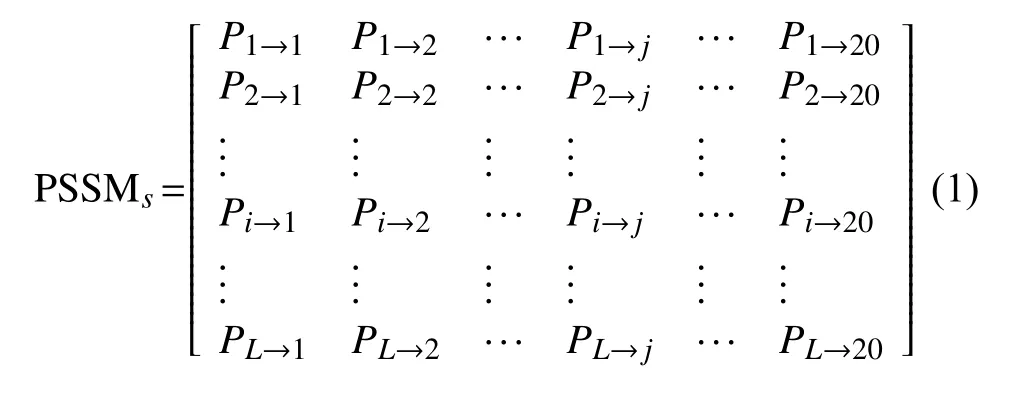
PSSM矩陣的每一行代表在查詢序列的相應位置發生在氨基酸替代的對數似然得分.位于矩陣第i行第j列的元素Pij表示在進化過程中查詢序列的第i個位置的氨基酸突變成j類氨基酸的得分.
1.2 二級結構劃分標準
蛋白質二級結構通常分為8類:G(310-helix),H(αhelix),I(π-helix),B(isolated β-bridge),E(β-stand),S(bend),T(hydrogen bonded turn)和rest(apparently random conformations).主流的PSSP思想會將這8類結構歸納為3種構象(H、E和C).通常情況下,H、E和C三種構象之間沒有明確的界限而且也沒有統一的標準去劃分這三種構象.然而在1999年,Cuff和Barton兩位學者證實了劃分方案可以影響最后的預測精度,所以人們希望找到一種劃分方案可以獲得更高的預測精度.由此,蛋白質二級結構字典法(DSSP[17])獲得了廣泛的認可.該方法依據已知的氫鍵相連的部分劃分二級結構.本文選用DSSP方法,將8類結構明確歸納為:H、G屬于Helices,記作H;E、B屬于Sheets,記作E;G、S、T、C、I屬于Coils,記作C.
2 基于多重進化矩陣的特征向量構造方法
在實際中,蛋白質的結構是不斷折疊式的,某個殘基不僅與它相鄰的殘基發生作用,還可能與它在序列上相差較遠的某些殘基發生作用,且蛋白質進化過程中氨基酸位點存在突變的可能.PAM矩陣和BLOSUM矩陣就反映了蛋白質中存在的氨基酸突變.
因PAM矩陣和BLOSUM矩陣都是PSI-BLAST程序中的打分標準,不同的打分矩陣對于評價氨基酸突變是不同的[18,19],例如PAM250矩陣假設每100個氨基酸發生250次點突變,PAM矩陣存在從PAM1到PAM250的情況.由于PAM矩陣是基于近相關蛋白比對得到的打分矩陣,BLOSUM矩陣是基于觀測到的遠相關蛋白對比得到的打分矩陣.本文參考了PAM矩陣和BLOSUM矩陣之間的相互關系[20],如圖1所示,設計了多重進化矩陣編碼方式.為更詳細的描述氨基酸位點發生突變的可能性和序列內部近距離和遠距離的氨基酸之間的相互影響,選擇了低趨異度矩陣PAM30和高趨異度矩陣PAM250和BLOSUM62矩陣三種不同趨異度的進化矩陣來表達蛋白質序列.
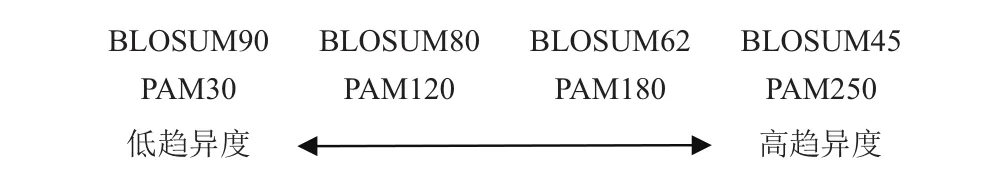
圖1 PAM矩陣和BLOSUM矩陣概要
首先將蛋白質序列送入PSI-BLAST程序,通過調整參數,得到廣泛使用的BLOSUM62矩陣、低趨異度矩陣BLOSUM90和高趨異度矩陣PAM250.將得到的三種不同趨異度的進化矩陣對齊特征維度,組合得到60維的向量表示原來的蛋白質序列,考慮臨近殘基的影響,采用滑動窗口法對所得特征向量進行處理,設置滑動窗口為13,得到一個780維向量表示原來的序列,構成多重進化矩陣特征.
為了能夠用計算方法進行訓練和預測,需要將相差較大的原始值進行規范化處理.本文利用公式(2)把多重進化矩陣的元素標準化到0~1之間.

其中x是多重進化矩陣中元素的原始值.
3 蛋白質二級結構預測框架
為了在構造特征向量時能更好的反映蛋白質序列中氨基酸殘基存在突變的可能性,且考慮預測過程中存在分類器參數選擇困難及可靠性差等問題,本文提出基于多重進化矩陣的蛋白質二級結構預測方法,其具體過程如下:
1)首先要將BLAST本地化.下載蛋白質NR數據庫及BLAST程序本地軟件包,對BLAST進行本地配置.
2)計算蛋白質序列的位置特異性打分矩陣(PSSM)矩陣,設置PSI-BLAST程序的參數為(-num_iterations:3,-eavlue:0.001,-matrix:BLOSUM62),得到該參數條件下的PSSM矩陣.
3)調整PSI-BLAST程序參數,將matrix分別設置為BLOSUM90和PAM250,計算該參數條件下的PSSM矩陣.
4)將3)中得到的三種進化矩陣對齊特征維度,組合得到60維的向量來表示原來的蛋白質序列.采用滑動窗口法處理向量,設置滑動窗口為13,得到一個780維向量來表示原來的蛋白質,構成多重進化矩陣特征,對矩陣進行標準化處理.
5)利用網格搜索法和K折交叉驗證來優選實驗參數.選取強分類器多分類支持向量機M-SVMCS來說明實驗過程:
① 設定網格搜索的變量(c,p)的范圍以及搜索步距,選擇使分類準確率最高的一組c和p;
② 在尋得了局部最優參數之后,再在這組參數附近選擇一個小區間,采用小步距進行二次精搜,再次選擇使分類準確率最高的一組c和p;
③ 涉及的所有參數對都用7折交叉驗證進行實驗,按數據集條數平均分成7份,每次選擇其中6份做訓練集,剩下的1份做測試集,重復7次;
④ 上述提到的分類準確率的參數對按照以下原則確定:若參數選擇過程中有多組c和p對應于最高的驗證分類準確率,則選取能夠達到最高驗證分類準確率中參數c最小的那組c和p作為最佳的參數;如果對應最小的c有多組p,就選取搜索到的第一組c和p作為最佳參數對;
6)按照5)中獲得的最優參數模型,輸入結構未知的蛋白質序列特征,預測各個位點殘基二級結構.
4 實驗與結果分析
4.1 數據集
為了檢驗模型的預測精確性,選擇數據集要慎重,需要結合機器學習和生物學方面的知識.伴隨著PDB等主要蛋白質結構數據庫中的蛋白質結構資源的日益豐富,可用的蛋白質二級結構預測的樣本也越來越多.出于對實驗結果的公平及公正性的考慮,本文選擇三個廣泛應用的低同源性數據集RS126[21]、CB513[22]和25PDB[23]作為本文的實驗數據集,序列相似性均低于25%.RS126數據集含有126條非同源蛋白質序列.CB513數據集含有513條非同源蛋白質序列.25PDB數據集含有 1673條非同源蛋白質和從PDB中下載和掃描的高分辨率結構域.
4.2 參數選擇
為了證明本文提出的多重進化矩陣是一種有效特征向量表示方法,本文選擇了兩種弱分類器Logistics、RandomForest和一種強分類器MSVMpack進行實驗.其中Logistics和RandomForest來自WEKA軟件,MSVMCS來自MSVMpack軟件[24].三種分類器都是通過網格搜索法來挑選實驗參數.為了對分類器參數進行優化,且保證優化結果的可靠性,本方法結合七折交叉驗證與網格搜索法來確定實驗參數.
經過多組實驗,對所獲得的實驗結果進行對比,選擇其中最好的一組作為最優參數.對于數據集CB513和25PDB我們將針對不同分類算法得到的最優參數匯總如表1所示.
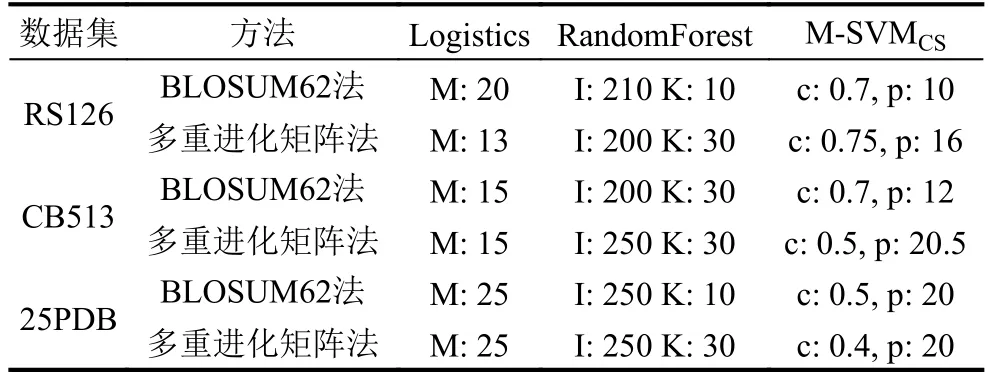
表1 最優參數結果表
4.3 結果評價標準
關于蛋白質二級結構預測結果的評價標準有很多種.目前在國際上大多使用以下幾種標準:
(1)整體預測準確率Q3
目前應用最廣泛的準確率,它指的是被正確預測的3種二級結構(殘基)的總百分比,可由公式(3)計算得出.
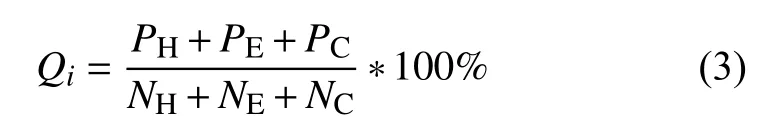
其中,NH、NE和NC分別表示序列中二級結構為H、E和C的殘基的總個數,PH、PE和PC分別表示被正確預測為H、E和C構象的殘基個數.
(2)三態預測準確率Qi
我們用Qi來表示每種二級結構被正確預測為H,E或C構象的預測準確率.可由公式(4)計算得出:
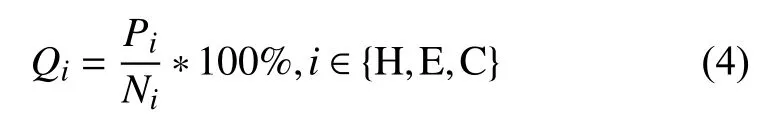
其中,Pi是待預測序列中被正確預測的處于i構象的殘基數目,Ni是待預測序列中被正確預測的處于i構象的殘基數目,i屬于H構象、E構象或C構象.
根據本文第4節的方法,我們在RS126、CB513和25PDB數據集上進行實驗.在RS126數據集上三個獨立分類器得到的整體預測準確率分別為67.86%、67.90%和73.90%,其各項獨立指標如表2所示.在CB513數據集上三個獨立分類器得到的整體預測準確率分別為65.53%、71.32%和75.50%,其各項獨立指標如表3所示.在25PDB數據集上三個獨立分類器得到的整體預測準確率分別為68.57%、72.62%和76.72%,其各項獨立指標如表4所示.

表2 RS126數據集使用BLOSUM62矩陣預測結果(%)
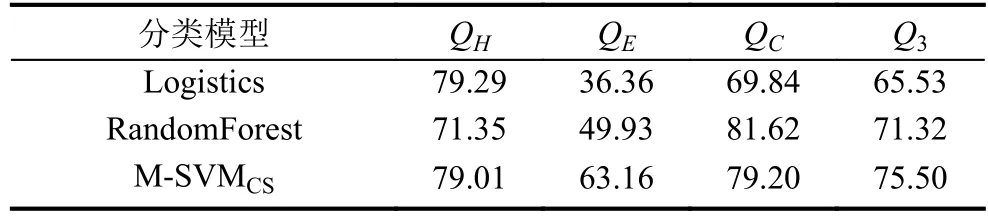
表3 CB513數據集使用BLOSUM62矩陣預測結果(%)
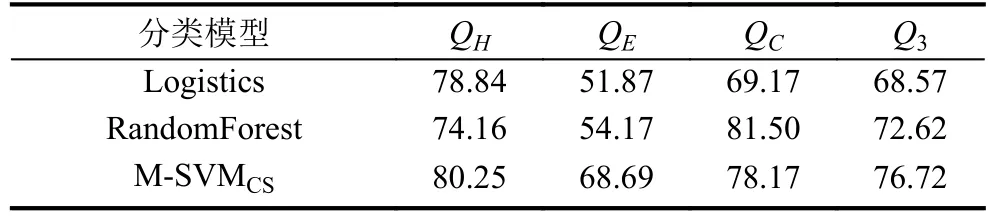
表4 25PDB數據集使用BLOSUM62矩陣預測結果(%)
然后,我們組合三種不同進化趨異度的矩陣,作為三個獨立分類器的輸入向量,通過網格搜索法和7折交叉法優選實驗參數,獲得優化參數模型,輸入結構未知的蛋白質序列特征,預測各個位點殘基二級結構.對數據集RS126使用三種分類器獲得的整體預測準確率分別是66.40%、68.08%和74.05%,各類別的預測準確率如表5所示.對數據集CB513使用三種分類器獲得的整體預測準確率分別是69.18%、71.89%和75.92%,各類別的預測準確率如表6所示.通過對比表2和表5可以看出,相比于傳統的實驗方法,多重進化矩陣這種表示方法在RS126數據集上分別高出了-1.37%、0.18%和0.15%.通過對比表3和表6可以看出,相比于傳統的實驗方法,多重進化矩陣這種表示方法在CB513數據集上分別高出了3.65%、0.57%和0.42%.而對于數據集25PDB得到的整體預測準確率分別是70.57%、73.16%和78.05%,各類別的預測準確率如表5所示.通過對比表4和表7可以看出,相比于傳統的實驗方法,多重進化矩陣這種表示方法在25PDB數據集上分別高出了2.00%、0.54%和1.33%.各表中整體預測準確率提高的值用粗體顯示.
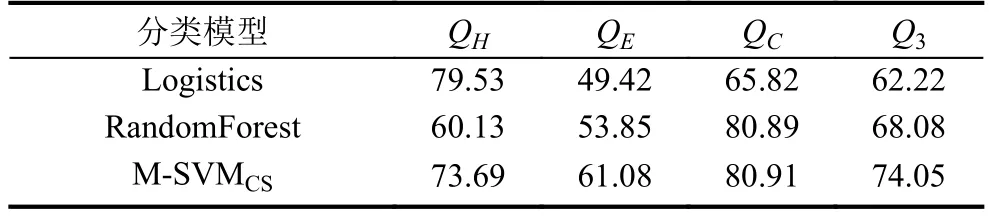
表5 RS126數據集使用多重進化矩陣預測結果(%)
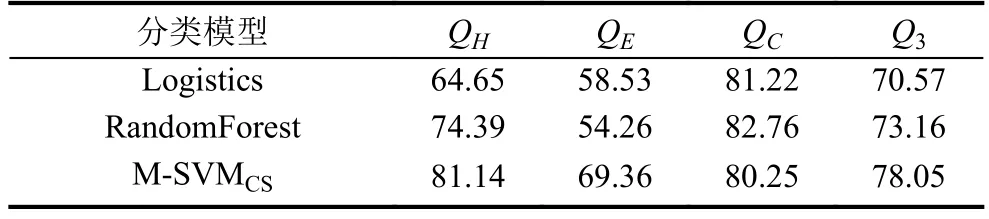
表7 25PDB數據集使用多重進化矩陣預測結果(%)
為了更為直觀的體現本文方法的有效性,本文將在RS126數據集、CB513數據集和25PDB數據集上使用不同算法得到的整體預測準確率表示成圖2、圖3和圖4,從中可以看出,本方法相對于原BLOSUM62蛋白質序列表示方法,除去對RS126數據集使用Logistics分類器得到的結果有所下降,在其他對比實驗中得到的整體預測準確率均有提高.對于這種現象,由于:
(1)相對于CB513數據集和25PDB數據集,RS126數據集數據量比較少,包含的蛋白質種類少 .
(2)Logistics分類器的分類精度受樣本數據量的影響,當樣本數量較小時,結果存在的風險較大.
綜合這兩種因素,我們認為,對于邏輯回歸分類器,使用多重進化矩陣反而分類精度有所下降這種現象是正常的,不能否認多重進化矩陣是一種有效的蛋白質序列特征表示方法.我們將對于三個數據集的使用MSVMCS分類器得到的結果匯總,如圖5所示.從表2至表7和圖5可以看出,在整體預測準確率上,本文方法比BLOSUM62矩陣表示方法在不同數據集上分別提高了 0.15%、0.42%和1.33%,對于數據集RS126提升較小,對于25PDB數據集提升較大.說明多分類支持向量機比較適用于大樣本數據集,而對于小樣本數據集效果并不明顯.
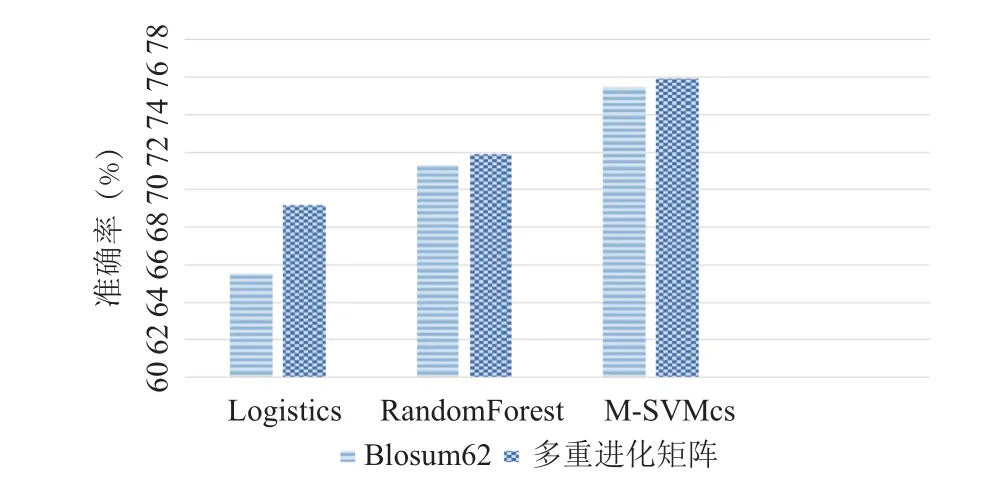
圖2 不同方法在數據集RS126的整體預測準確率
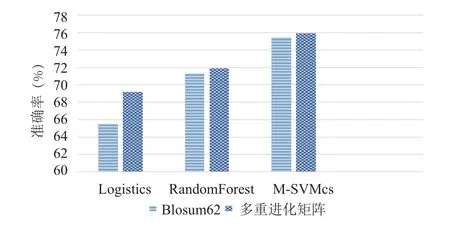
圖3 不同方法在數據集CB513的整體預測準確率
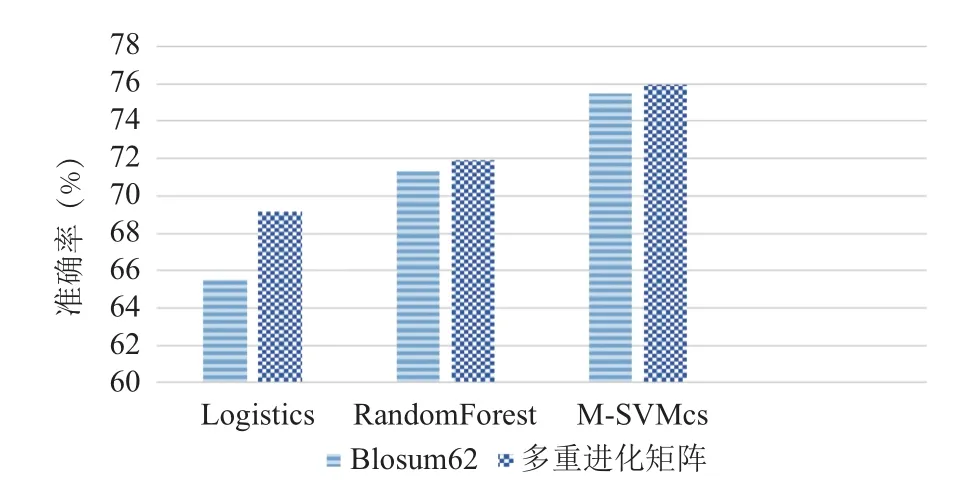
圖4 不同方法在數據集25PDB的整體預測準確率
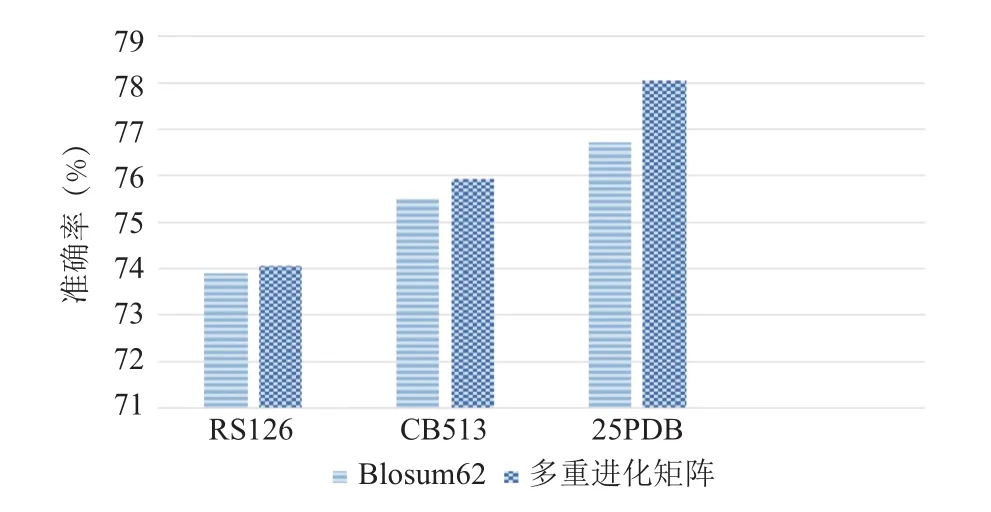
圖5 M-SVMCS分類器在不同數據集的整體預測準確率
5 結語
本文根據蛋白質序列不同進化趨異度之間的關系,組合PAM矩陣和BLOSUM矩陣,設計了一種新的方法來構成特征向量表示蛋白質序列信息;選用Logistics、RandomForest和M-SVMCS機器學習模型作為預測工具,采用交叉驗證法和網格搜索法相結合來確定實驗參數,預測各個位點殘基二級結構.在數據集RS126、CB513和25PDB上開展的對比實驗,表明本文所提出基于多重進化矩陣的蛋白質特征向量構造方法能夠有效提高蛋白質二級結構的預測精度.
在下一步的工作中,我們可以從下面幾點做出改進:(1)深入研究蛋白質信息特征提取算法,加入對蛋白質二級結構特征信息的描述;(2)嘗試利用特征選擇算法優選特征,降低特征向量維度,提高分類器計算速度.在分類算法上進行可能的改進也是下一步研究的重點.
1Jones DT.Protein structure prediction in the postgenomic era.Current Opinion in Structural Biology,2000,10(3):371-379.[doi:10.1016/S0959-440X(00)00099-3]
2澤瓦勒貝,鮑姆.理解生物信息學.李亦學,郝沛,譯.北京:科學出版社,2012.
3Floudas CA.Computational methods in protein structure prediction.Biotechnology and Bioengineering,2007,97(2):207-213.[doi:10.1002/(ISSN)1097-0290]
4Khoury GA,Smadbeck J,Kieslich CA,et al.Protein folding and de novo protein design for biotechnological applications.Trends Biotechnology,2014,32(2):99 -109.[doi:10.1016/j.tibtech2013.10.008]
5張海霞,唐煥文,張立震,等.蛋白質二級結構預測方法的評價.計算機與應用化學,2003,20(6):735-740.
6Lee D,Redfern O,Orengo C.Predicting protein function from sequence and structure.Nature Reviews Molecular Cell Biology,2007,8(12):995-1005.[doi:10.1038/nrm2281]
7Cai YD,Zhou GP.Prediction of protein structural classes by neural network.Biochimie,2000,82(8):783-785.[doi:10..1016/S0300-9084(00)01161-5]
8Mandle AK,Jain P,Shrivastava SK.Protein structure prediction using support vector machine.International Journal on Soft Computing,2012,3(1):67-78.[doi:10.5121/ijsc]
9Wang S,Peng J,Ma JZ,et al.Protein secondary structure prediction using deep convolutional neural fields.Scientific Reports,2016,6:18962.[doi:10.1038/srep18962]
10Lu Z,Szafron D,Greiner R,et al.Predicting subcellular localization of proteins using machine-learned classifiers.Bioinformatics,2004,20(4):547-556.[doi:10.1093/bioinformatics/btg447]
11Chou KC,Cai YD.Predicting protein localization in budding yeast.Bioinformatics,2005,21(7):944-950.[doi:10.1093/bioinformatics/bti104]
12Cai YD,Chou KC.Predicting 22 protein localizations in budding yeast.Biochemical and Biophysical Research Communications,2004,323(2):425-428.[doi:10.1016/j.bbrc.2004.08.113]
13Wang JR,Wang C,Cao JJ,et al.Prediction of protein structural classes for low-similarity sequences using reduced PSSM and position-based secondary structural features.Gene,2015,554(2):241-248.[doi:10.1016/j.gene.2014.10.037]
14梅娟,趙吉,傅毅.基于圖聚類和序列信息的蛋白質遠同源性探測.計算機與應用化學,2015,32(8):945-950.
15Wang L,You ZH,Xia SX,et al.Advancing the prediction accuracy of protein-protein interactions by utilizing evolutionary information from position-specific scoring matrix and ensemble classifier.Journal of Theoretical Biology,2017,418:105-110.[doi:10.1016/j.jtbi.2017.01.003]
16Ben-Gal I,Shani A,Gohr A,et al.Identification of transcription factor binding sites with variable-order Bayesian networks.Bioinformatics, 2005, 21(11):2657-2666.[doi:10.1093/bioinformatics/bti410]
17Sebastiani F.Text categorization.Rivero LC,Doorn JH,Ferraggine VE.Encyclopedia of Database Technologies and Applications.Hershey,US:Idea Group Reference,2005.683-687.
18Ortu?o FM,Valenzuela O,Prieto B,et al.Comparing different machine learning and mathematical regression models to evaluate multiple sequence alignments.Neurocomputing,2015,164:123-136.[doi:10.1016/j.neucom.2015.01.080]
19Lal D,Verma M.Large-scale sequence comparison.Keith JM.Bioinformatics:Volume I:Data,Sequence Analysis,and Evolution.New York:Springer,2017.191-224.
20喬納森·佩夫斯納.生物信息學與功能基因組學.孫之榮,譯.北京:化學工業出版社,2006.
21Rost B,Sander C.Prediction of protein secondary structure at better than 70% accuracy.Journal of Molecular Biology,1993,232(2):584-599.[doi:10.1006/jmbi.1993.1413]
22Cuff JA,Barton GJ.Evaluation and improvement of multiple sequence methods for protein secondary structure prediction.Proteins Structure Function and Bioinformatics,1999,34(4):508-519.[doi:10.1002/(ISSN)1097-0134]
23Kurgan LA,Homaeian L.Prediction of structural classes for protein sequences and domains-impact of prediction algorithms,sequence representation and homology,and test procedures on accuracy.Pattern Recognition,2006,39(12):2323-2343.[doi:10.1016/j.patcog.2006.02.014]
24http://www.Loria.fr/lauer/MSVMpack.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
