植物基因家族全基因組分析平臺構建及在Dof中的應用
2018-03-07 01:54:44王培培劉長寧
綠色科技 2018年13期
王培培 劉長寧


摘要:指出了基因家族是植物基因組的重要組成部分,在植物整個生長發育過程中扮演著重要角色,參與各種生物、非生物脅迫的應答反應,而且在植物環境適應性進化過程中可發揮重要作用。植物基因家族分析方法多樣,可選擇軟件較多,參數設置繁瑣,分析流程缺少規范性。為拉低學科差異的鴻溝與大數據分析的繁瑣性,構建了一個植物基因家族全基因組鑒定與分析平臺,以小桐子Dof轉錄因子基因家族全基因組分析為例進行了平臺測試,可為植物基因家族的研究提供一個流程化參考。
關鍵詞:基因家族;分析平臺;全基因組分析
中圖分類號:S432.2+3
文獻標識碼:A
文章編號:1674-9944(2018)07-0001-05
1 研究概況
由于存在物種特異性擴張,高等生物基因組中有豐富的多基因家族及超基因家族(superfamilies),20世紀70年代開始,人們逐漸對這種在生物個體和群體中產生遺傳變異并與基因組冗余有微妙關系的基因家族的研究產生了興趣[1]。基因家族成員以簇狀或無規律形式分布于基因組的不同位置并在植物生長發育過程中發揮重要作用。轉錄因子(transcription factor,TF)是基因表達的轉錄調控因子,調控復雜的DNA到RNA的時空特異性表達,轉錄因子基因家族是植物中最大、作用最廣泛的基因家族之一[2]。由轉錄因子組成的轉錄起始前復合物可以偶聯靶基因啟動子中的順式作用元件并啟動基因轉錄。例如,轉錄因子通過招募啟動子共激活因子、一般轉錄因子、染色質重塑因子等蛋白因子構成轉錄起始前復合物并激活RNA聚合酶,促進RNA轉錄鏈的起始及延伸[3]。
伴隨著二代測序的廣泛應用及生物信息學的快速發展,解析大數據密碼所隱含的生物學現象是生命科學研究者要解決的首要問題.基于這樣的契機,流程化分析平臺應運而生。一些不斷更新的分子生物學數據庫作為基因組序列的倉儲,為在全基因組水平研究轉錄因子基因家族提供了便利,常用的比較全面的大型數據庫比如NCBI、Ensembl和DDBJ等是轉錄因子研究的重要數據來源。常用的轉錄因子數據庫包括PlantTFDB4.0( http: //planttfdb.cbi.pku.edu.cn/)和PlnTFDB 3.0( http://plntfdb.bio.unipotsdam.de/v3.0/)等,為植物轉錄因子的研究提供了豐富的開源數據。
目前,有關植物基因家族全基因組分析的研究大部分都集中在一些重要的轉錄因子上,比如bZIP、MADS- box、SBP - box、WRKY、AP2/ERP、NAC等等,而對Dof( DNA- binding with one zinc finger)鋅指蛋白的報道相對較少,它是植物特有的一類轉錄因子基因家族,屬于鋅指蛋白超家族( zine finger super - family)。Dof在多種代謝途徑及植物生長發育過程中發揮著重要作用,包括C、N代謝、光響應、種子發育和萌發等[4]。首個Dof轉錄因子蛋白被發現于玉米中(ZmDofl),在玉米糊粉層形成過程中有重要功能朝。近年來,相繼從擬南芥[6]、水稻[7]、大麥[8]、小麥[9]、大豆[10]、蓖麻[11]等物種中鑒定出Dof基因,并對其功能進行了深入地研究,不斷證實了Dof基因的功能重要性。
各種模式生物基因組測序工作完成之后,基因家族成為功能基因組學的研究熱點之一。基因家族全基因組分析的專業人士可以根據數據特征及分析目的自主選擇分析策略。但是,對于非專業的基因家族研究者而言,目前缺少一個流程化的分析平臺。基于分子生物學和生物信息學研究背景的復雜性,搭建一個基因家族分析平臺有其重要的現實意義。目前關于基因家族分析有很多值得借鑒的軟件工具與操作方法,但仍然存在很多值得完善的地方。例如,分析方法多樣化,可選擇軟件較多,參數設置繁復,分析流程復雜等。研究參考已有的分析方法,利用現有的硬件設施選擇合適的分析工具初步搭建了植物基因家族全基因組分析平臺,為植物基因家族流程化、規范化分析提供參考。
2 平臺搭建
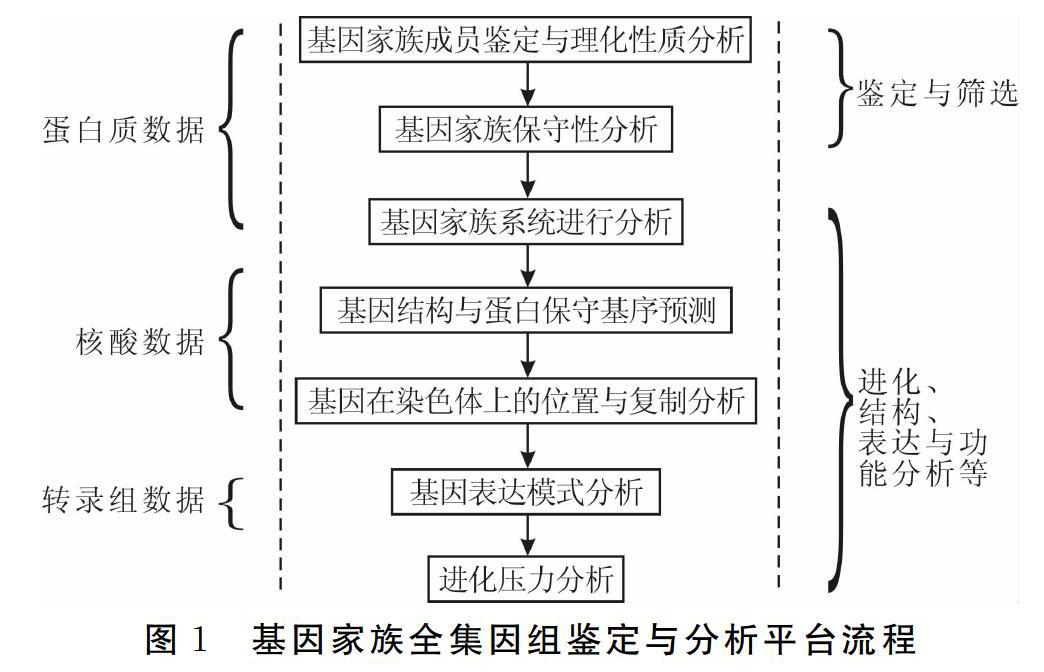
輸入并整合分析蛋白質數據、核酸數據、轉錄組數據,結合基因家族全基因組鑒定模塊與挖掘分析模塊,完成流程化基因家族全基因組鑒定與分析。
2.1 測試數據來源
平臺應用測試所涉及到的物種基因組信息是 從以下數據倉儲網站下載:小桐子最完整版基因組數據(Assembly JatCur1.0)來源于NCBI( https://WWW.ncbi. nlm. nih. gov/);擬南芥Dof基因家族蛋白質序列與核酸序列數據來源:TAIR 9.0(http://www. arabi-dopsis. org/);蓖麻Dof基因家族蛋白質序列與核酸序列數據來源:PlantTFDB database( http://planttfdb.cbi. pku. edu.cn/);小桐子Dof基因表達量數據收集于:SRA數據庫(https://www. ncbi. nlm. nih. gov/)。
2.2 平臺流程
平臺分析流程主要包括以下幾個步驟:基因家族成員鑒定與理化性質分析;基因家族保守性分析;基因家族系統進化分析;基因結構與蛋白保守基序分析;基因在染色體上的位置與復制分析;基因表達模式分析;進化壓力分析(圖1)。分析流程中參考已有的分析策略結合現有的硬件設備,選擇了一系列較為高效便捷的軟件工具(圖2)。
2.3 結構模塊
平臺沒計主要包括三個模塊,分別為可視化模塊、邏輯操作模塊和數據服務模塊。其中可視化用戶服務界面主要通過snakemake實現;邏輯操作模塊主要包含各個分析模塊及其涉及到的應用軟件T具;數據服務模塊主要是指基因家族分析使用的公共數據庫中的基因組數據(圖2)。
3 方法步驟
3.1 基因家族成員鑒定
為篩選出某物種基因家族所有成員,結合Blastp和hmmsearch兩種程序對全基因組數據進行全面搜索。首先,利用待鑒定物種的全基因組蛋白質數據,構建本地Blast數據庫,以模式植物擬南芥轉錄因子蛋白質作為query序列執行本地blastp程序(e- value設置為le-10)。其次,以Pfam蛋白結構域模型作為hm-mquery序列,以物種全基因組數據作為HMM數據庫,執行本地hmmsearch程序。兩部分篩選結果取交集,刪除冗余,所得候選序列利用SMART及NCBI- CDD工具進行蛋白質結構預測[12,13],刪除不含目標轉錄因子特定功能結構域的序列,同時剔除不含完整讀碼框的序列。利用ExPASy Proteomics Server(http://ex-pasy. org/)工具對所有目標轉錄因子蛋白氨基酸序列進行分子量、蛋白質長度以及等電點等理化性質預測分析[14]。
3.2 蛋白質保守性分析
為可視化分析目標轉錄因子蛋白的保守性,首先,使用DNAMAN軟件來提取目標基因家族蛋白質的保守區域,結合smart驗證目標蛋白是否含有該家族特定的保守的功能domain[15];其次,通過ClustalW軟件對目標基因家族成員進行多序列聯配比對分析,鑒定出高度保守的蛋白質結構域.找到標志性功能位點,并用同種顏色標示保守的氨基酸[16]。
3.3 系統演化分析
首先,利用guidance2 工具對蛋白質序列進行多序列聯配比對分析,設置梯度參數獲得信任值比較高的columns[17]。其次,使用提取得到的蛋白質序列,結合MEGA6 [18]軟件采用鄰接法(Neighbor - Joining NJ)生成目標基因家族的系統演化樹,替換模式選用“poissonmodel”,校驗參數Bootstrap設置為1000。最后,使用在線的進化樹美化軟件EvolView等軟件對系統演化樹進行二次編輯修飾[19]。
3.4 基因家族蛋白結構和功能基序的預測
每個成員基因核酸序列與核酸序列對應的CDS序列,提交到Gene Structure Display Server( GSDS2.0:http://gsds2. cbi. pku. edu. cn/)[20]軟件分析基因結構組成模式,包括內含子、外顯子分布模式和數量特征等;利用在線工具MEME (multiple expectation maximiza-tion for motif elicitation)[21]對轉錄因子蛋白的功能mo-tif進行預測分析,長度參數設定為5~150,預測數量設置為10。
3.5 染色體定位和基因復制分析
結合物種基因組注釋GFF3文件,提取目標基因在染色體上的位置信息,將所有目標基因定位在該物種的染色體上,通過MapInspect[22]繪圖軟件繪制目標基因家族基因組染色體定位圖。利用McScanX[23]軟件判定基因發生片段復制,軟件執行物種all-against-allblastp文件和包含基因位置信息的GFF3文件.計算該物種基因組中的共線性區段(e-value=le-10).發生于同一個共線性區段的目的基因對被認為是發生片段復制的基因對;另外,基因發生串聯復制事件的判定條件為:①兩個基因序列相匹配部分的長度大于較長序列的80%;②兩個基因序列相匹配部分的相似性應大于80%;③緊密相連的基因中,只參與一次復制事件。結合基因在染色體上的位置,兩個基因應位于同一條染色體上[24]。
3.6 選擇壓力分析
每一對發生復制的基因,根據其CDS序列,利用DnaSP[25]軟件計算復制基因對的非同義替換率(Ka)和同義替換率(Ks)以及Ka/Ks值.分析發生復制事件的基因所受到的環境選擇壓力。①若Ka/Ks>1,正選擇壓力;②若Ka/Ks=1,受到中性選擇或自然選擇壓力;③若Ka/Ks<1,存在純化選擇作用。
3.7 基因家族的表達模式分析
基因的差異表達模式分析是基因功能研究的重要方法,為了進一步探究目標轉錄因子基因功能,收集基因在不同環境條件下(例如激素處理、鹽脅迫、干旱脅迫等)以及不同組織器官中的表達量數據(RNA - seq數據、表達芯片數據等),基因表達數據進行標準化處理之后,利用R、Heml[26]等軟件繪制表達譜熱圖。
4 測試與應用
鑒于植物基因家族分析缺少流程化、規范化平臺,構建了一個初級的基因家族全基因組鑒定與分析的平臺,依據其具體流程步驟,以小桐子Dof基因家族的全基因組鑒定與分析為例,對該平臺的方法進行實踐測試。小桐子是大戟科重要的多年生木本植物,因其種子含油量較高及花發育過程的特殊性是大戟科研究的一個潛在的模式植物。平臺測試分析過程中,對小桐子Dof基因進行全基因組篩選與鑒定,并對鑒定到的基因進行理化性質、保守性、基因進化、基因結構及功能mo-tif、染色體定位、表達譜、選擇壓力等進行系統性地研究分析,為小桐子Dof基因后續功能研究與開發利用提供理論基礎(表1)。
一共鑒定出了24個Dof基因,共編碼33條Dof蛋白,均屬于大分子蛋白,其理化性質預測結果列表如表2。
5 總結
越來越多的植物全基因組測序完成,對基因家族的研究也越來越普遍,本研究初步搭建了一個植物基因組分析平臺。主要成果包括:整理了基因家族分析的比較常見的分析方法,歸納出了主要分析流程;設計了分析平臺主要結構模塊主要包括可視化層一邏輯操作層一數據服務層等主要框架。利用小桐子Dof基因的流程化分析對該植物基因家族分析平臺進行了測試應用,驗證了平臺的可行性。隨著高通量測序的大量產出和發布,本課題的研究工作能為從事植物基因家族分析的工作者提供參考,輔助其完成不同目的的基因家族分析。此外,本課題的工作對相關生物信息學平臺的設計與構建也有一定的參考價值。
6 展望
目前,由于時間的限制、技術的不成熟等原因,該分析平臺仍然存在很多值得完善的地方。比如:本研究平臺初步建立,經驗不足.有待繼續完善;自動化程度較低,后續snakemake可視化分析需要完善;本平臺可以為基因家族功能性分析及其分析平臺設計提供一定的參考。
參考文獻:
[1]Hool, Campbellj, Elglns. The organization, expression, and evo-lution of antibody genes and other multigene families [J]. Annualreview of genetics, 1975, 9(1): 305~53.
[2] Van Lijsebettensm, Gasser K D. Transcript elongation factors:shaping transcriptomes after transcript initiation [J]. Trends inplant science, 2014, 19(11) : 717~26.
[3] Hahn S, Young E T. Transcriptional regulation in Saccharomy-ces cerevisiae: transcription factor regulation and function, mech-anisms of initiation, and roles of activators and coactivators [J].Genetics, 2011, 189( 3) : 705.
[4] Lijavetzky D, Carbonero P, Vicente- Carbajosa J. Genome-wide comparative phylogenetic analysis of the rice and ArabidopsisDof gene families [J]. BMC evolutionary biology, 2003, 3
[5] YaNngisawa S, Sheen J. Involvement of maize Dof zinc fingerproteins in tissue - specific and light - regulated gene expression[J]. The Plant Cell, 1998, 10(1): 75~89.
[6] Riechmann J L, Heard J, Martin G, et al. Arabidopsis transcrip-tion factors: genome - wide comparative analysis among eu-karyotes [J]. Science, 2000, 290(5499) : 2105.
[7] Lihavetaky D, Carbonero P, Vicente- Catbajosa J. Genome-wide comparative phylogenetic analysis of the rice and ArabidopsisDof gene families [J]. BMC evolutionary biology, 2003, 3
[8] Moreno-Risueno M , Mart Nez M, Vicente- Carbajosa J, et al.The family of DOF transcription factors : from green unicellular al-gae to vascular plants [J]. Molecular Genetics and Genomics,2007, 277(4) : 379.
[9] Chen ay, Guo xj, Chen zx, et al. Genome- wide characterizationof developmental stage- and tissue- specific transcription factorsin wheat [J]. BMC genomics, 2015, 16(1) : 125.
[10] Wang H W, Zhang B, Hao Y J, et al. The soybean Dof - typetranscription factor genes, GmDof4 and GmDofll, enhance lipidcontent in the seeds of transgenic Arabidopsis plants [J]. ThePlant Journal, 2007, 52(4) : 716.
[11] Jin Z, Chandrasekaran U, Liu A. Genome- wide analysis of theDof transcription factors in castor bean ( Ricinus communis L. )[J]. Genes &- genomics, 2014, 36(4): 527.
[12] Xu Q, Dunbrack R L. Assignment of protein sequences to exist-ing domain and family classification systems: Pfam and the PDB[J]. Bioinformatics, 2012, 28(21) : 2763.
[13] Marchler- Bauer A, Zhenh C, Chitsaz F, et al. CDD: con-served domains and protein three- dimensional structure [J].Nucleic acids research, 2012(11).
[14] Artimo P, Jonnalagekka M, Arnold K, et al. ExPASy: SIBbioinformatics resource portal [J]. Nucleic acids research,2012, 40(W1) : 597~603.
[15] Woffelman C. DNAMAN for windows, version 5.2. 10: LynonBiosoft [J]. Institute of Molecular Plant Sciences, Netherlands:Leiden University, 2004(4).
[16] Fukami- Kobayashl K, Salto N. How to make good use ofCLUSTALW [J]. Tanpakushitsu kakusan koso Protein, nucleicacid, enzyme, 2002, 47(9): 1237.
[17] Sela I, Ashkenazy H, Katoh K, et al. GUIDANCE2: accuratedetection of unreliable alignment regions accounting for the un-certainty of multiple parameters [J]. Nucleic Acids Research,2015, 43(W1).
[18] Tamura K, Stecher G, Peterson D, et al. MEGA6: molecularevolutionary genetics analysis version 6.0 [J]. Molecular biolo-gy and evolution, 2013(8).
[19] Zhang H, Gao S, Lercher M J, et al. EvolView, an online toolfor visualizing, annotating and managing phylogenetic trees [J].Nucleic acids research, 2012, 40(W1) : 569.
[20] Hu B, Jin J, Guo AY, et al. GSDS 2.0: an upgraded gene fea-ture visualization server[J]. Bioinformatics, 2014, 31(8) : 1296.
[21] Bailey T L, Johnson J, Grant C E, et al. The MEME suite [J].Nucleic acids research, 2015, 43( Wl) : 39~49.
[22] He H, Dong Q, Shao Y, et al. Genome- wide survey and char-acterization of the WRKY gene family in Populus trichocarpa[J]. Plant Cell Reports, 2012, 31(7) : 1199.
[23] Wang Y, Tang H, Debarry J D, et al. MCScanX: a toolkit fordetection and evolutionary analysis of gene synteny and col-linearity [J]. Nucleic acids research, 2012, 40(7) : 49.
[24] Jin J, Tian F, Yang D-C, et al. PlantTFDB 4. 0: toward acentral hub for transcription factors and regulatory interactionsin plants [J]. Nucleic acids research, 2016(4).
[25] Librako P, Rozas J. DnaSP v5: a software for comprehensive a-nalysis of DNA polymorphism data [J]. Bioinformatics, 2009,25 (11) : 1451.
[26] Deng W, Wang Y, Liu Z, et al. Heml: a toolkit for illustratingheatmaps [J]. PloS one, 2014, 9(11).
