基于文獻挖掘視角的組學研究脈絡梳理
2018-03-21 05:09:56,,,,
中華醫學圖書情報雜志 2018年3期
關鍵詞:研究
,,, ,
隨著科學研究的進展,人們發現單純研究某一方向無法解釋全部生物醫學問題,科學家便提出從整體出發研究人類組織細胞結構、基因、蛋白及其分子間相互的作用,通過整體分析反映人體組織器官功能和代謝的狀態,因此便產生了“組學”的概念。從分子生物學角度,組學主要涵蓋基因組學、蛋白組學、代謝組學、轉錄組學、脂類組學、免疫組學、糖組學和 RNA組學等。Omics是組學的英文稱謂,其詞根“-ome”在英文中是指一些種類個體的系統集合。Genomics(基因組學)是最早提出的組學類型,由美國科學家Thomas Roderick于1986年提出[1],之后其他類型的組學相繼出現。筆者通過查閱分析國內外大量組學相關綜述后發現,現階段的組學研究綜述都是關注某一種組學的最新進展,缺少從宏觀角度分析多種組學的融合研究。就目前組學研究的態勢而言,多種組學技術融合已成為必然趨勢。因此,全面研究組學的整體發展趨勢和各類組學之間的脈絡關系,顯得十分重要。文本挖掘技術和信息計量學方法的發展為從海量的科研文獻中梳理組學研究脈絡提供了可能[2]。
文獻是科研成果的主要產出和表達形式,是由科研工作者對其創造性研究成果進行理論分析和科學總結并公開發表的文體,也是醫學事業不斷發展的重要科技信息源,是記錄醫學科技進步、重大發明和改革的歷史性文件[3]。文獻挖掘[4]是數據挖掘領域中一個重要研究方向,其處理對象是文本類型的文獻數據。一般通過統計方法獲取所關注的文獻,再使用自然語言處理方法從中抽取出特定的事實信息,并對內容進行分析,從非結構化的數據中分析出隱藏的一些規律。文獻挖掘方法已在多個領域中得到了廣泛的應用,如生物學、醫藥學、生物醫藥學以及科學計量學等。文獻挖掘技術[5]主要包括信息檢索、實體識別和信息抽取。實體識別[6]旨在發現文獻中重要的實體,該技術中常見的方法為基于特征、基于詞典或者基于規則進行實體識別。而信息抽取技術主要把文獻中含有的重要信息或者事實抽取出來,并用形式化的結構表示,依據共現關系[7]和自然語言處理技術[8]進行文本內容關系的抽取。
文獻計量分析[9]有助于全面了解某一研究領域的國內外文獻發表情況,目前以所有組學為對象的文獻計量分析少之又少。通過分析國內外文獻發表情況,方便該領域研究人員了解組學的研究現狀及發展方向,有助于科研管理機構在項目評審、資助中合理分配資源,有助于其科研選題、成果發表及選擇研究合作方并調整研究方向[10]。
本文擬利用文獻計量學方法,借助PubMed數據庫及相關文獻挖掘、分析方法對“組學(Omics)”相關英文文獻進行統計和分析,探尋組學的研究軌跡,為研究人員更加深入系統地開展組學研究提供參考。
1 資料來源與方法
本文使用的數據集來自PubMed數據庫[11]。美國國立生物技術信息中心(National Center for Biotechnology Information,NCBI)提供的The Entrez Programming Utilities(E-utilities)編程工具,是訪問NCBI Entrez查詢和PubMed數據庫的穩定接口,可以實現PubMed數據庫記錄的批量下載。本文使用E-utilities中的Esearch和Efetch 2種工具獲取PubMed記錄,時間跨度為1896-2016年,共獲得27 040 819條記錄,包含所有的出版類型。本文關注的主題為“組學(Omics)”。組學是研究一些種類個體的系統集合的學科,如基因組是構成生物體所有基因的組合,基因組學這門學科是研究這些基因以及這些基因間的關系,因此我們將組(Omes)與組學(Omics)同等對待。具有組學含義的單詞均有一個共同的特征,即以“-ome” “-omes”“-omic”或“-omics”結尾,故本文選取文獻的“title”或“abstract”為統計窗口,從中識別具備上述特征的單詞,在數據集中共識別出19 268個具備上述特征的單詞。通過刪除噪音單詞(如“some” “home”等),最后得到77個出現頻次不低于10次的“-Omics”單詞(表1),以供下一步分析。將上述77個“-Omics”單詞重新在原始數據的題目和摘要中用Python語言編寫的程序進行匹配,27 040 819條原始數據中含有77個“-Omics”單詞中的任何一個的記為有效數據,共得到346 977條記錄作為本文的數據集。
可視化分析采用VOSviewer,它是一款用來構建和查看文獻計量圖譜的免費文獻計量分析軟件,基于文獻的共引和共被引原理,可用于繪制各個知識領域的科學圖譜。將所有類型組學的共現數據經過處理后導入VOSviewer進行可視化,得到網絡可視化圖。圖中圓圈和標簽代表關鍵詞,圓圈及標簽大小代表其重要性的高低,擁有相同顏色的圓圈屬于同一個聚類[12]。
主題河是一種被證明為可有效反映文本之間的時間屬性的方法。在這種可視化方法中,時間被表示為從左往右的一條水平軸,然后用不同的顏色條帶代表不同的主題,條帶的寬度代表該主題在該時間的一個度量。這樣人們可以跟蹤任何一個主題在量上隨時間的變化,也能比較不同的主題在同一個時刻相對規模的大小[13]。
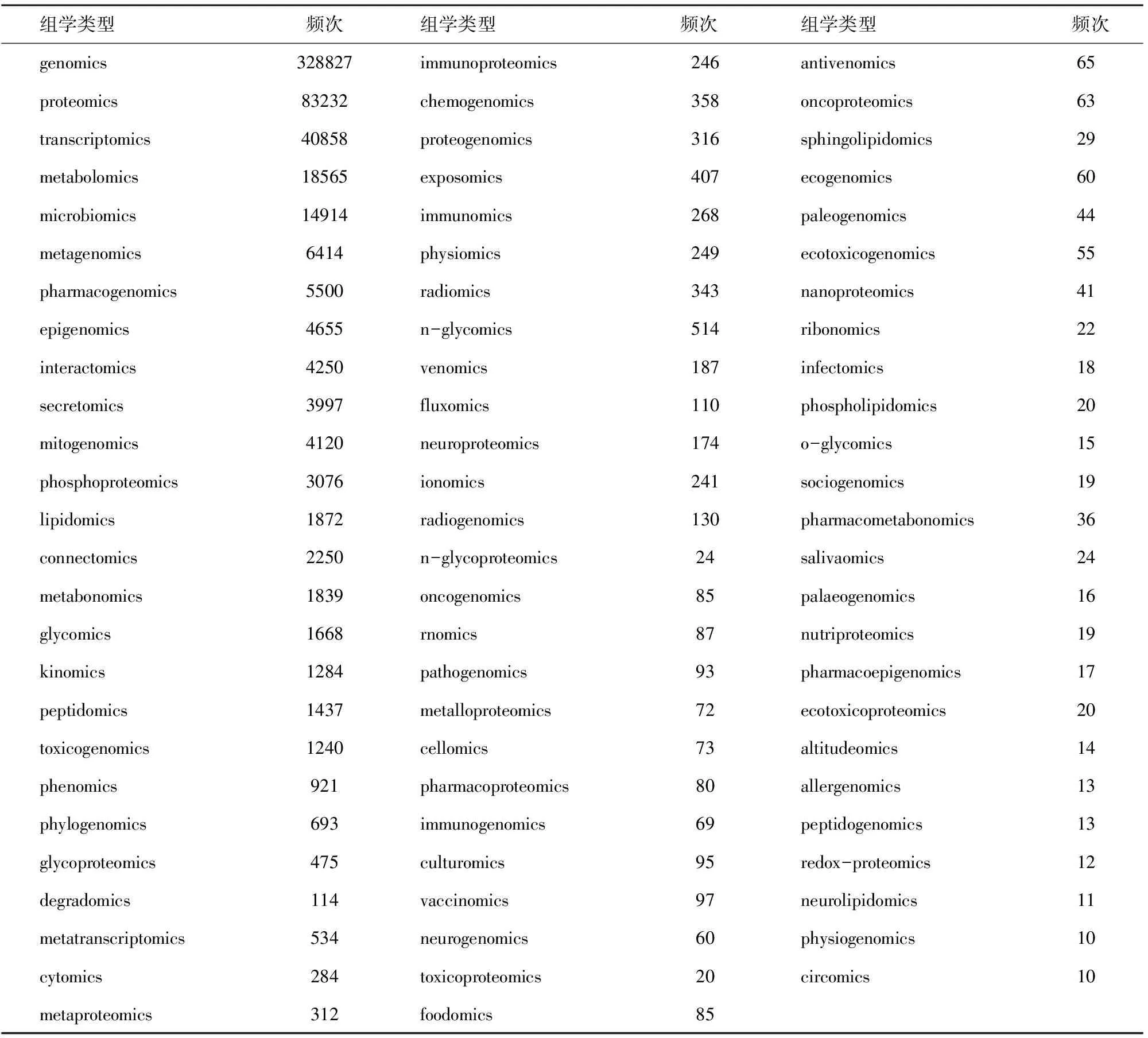
表1 出現頻次不低于10次的“-Omics”單詞
2 結果與分析
從上述數據集中篩選出關于組學的相關文獻共計346 977篇,包括期刊論文345 549篇(占99.59%)和綜述1 428篇(占0.41%)。
2.1 文獻年度變化趨勢
文獻的年度分布情況可以從一定程度上反映該領域的發展情況。分析文獻量與時間變化的關系可以反映研究主題的發展情況,可以大體揭示該主題的發展階段與規律。本文將組(-ome/-omes)與組學(-omic/-omics)同等對待,美國科學家Thomas Roderick于1986年最先提出的是Genomics(基因組學),而第一篇提到“基因組(genome)”的文獻則出現在1943年。1943-2016年全世界組學相關文獻發表情況如圖1所示。從1943年之后組學相關研究的發表量整體呈逐年遞增趨勢,從1999年的4 331篇迅速增長到2000年的5 288篇,到2016年文獻發表量已達40 590篇。
人類基因組計劃(Human Genome Project,HGP)由美國于1987年啟動, 2000年6月26日參加人類基因組工程項目的美國、英國、法國、德國、日本和中國等6國科學家共同宣布,人類基因組草圖的繪制工作已經完成,后基因組時代來臨。組學領域的研究文獻呈現了井噴式的增長,已有越來越多的國內外科學工作者投入到組學研究中,并獲得了大量的研究成果,組學已逐漸成為生物醫學研究領域的熱點之一。
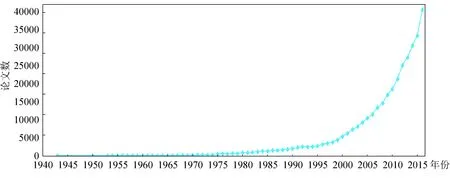
圖1 組學研究論文發表情況
2.2 各類組學文獻情況
2000年之前組學研究類型較單一,之后各類組學研究相繼涌現,并呈現出不同的變化。我們選取數據中論文總數排名前10的組學類型進行比較,發現各類組學都呈現了逐年遞增的現象。其中基因組學的文獻發表量遙遙領先,蛋白質組學和轉錄組學的文獻發表量緊隨其后。2000-2014年,蛋白質組學的文獻發表量一直高于轉錄組學,2014年之后轉錄組學的文獻發表量趕超了蛋白質組學。原因在于,從2008年開始,第二代測序技術利用一系列高通量測序技術(high throughput sequencing)進行大規模的基因組DNA或RNA測序,能夠快速準確地獲得基因組編碼序列,滿足極短時間內對基因組進行高分辨率檢測的要求。隨著第二代測序技術高通量、高準確率、低成本等優點的實現,轉錄組學測序技術也隨之得到了更廣泛的應用[14]。因此,轉錄組學的關注度逐漸升高并且超過了蛋白質組學的關注度。
2.3 各類組學共現情況
統計不同類型的“組學”之間在同一篇文獻的題目和摘要中出現的情況,便可形成多組學研究的相關關系。將多組學共現類型細分為在同一篇文獻中分別出現2種類型、3種類型、4種及4種以上類型,并進行分類計量。1995年首次出現多組學共現的文獻。2種類型組學共現的文獻量一直處于遙遙領先的狀態,3種類型組學共現和4種及4種以上組學類型共現的文獻量較2種類型組學共現的文獻量還有一些差距,但總體來說各種共現情況都隨著時間的增長呈現出逐年遞增的趨勢。
將多組學共現數據導入VOSviewer中,其結果以可視化圖譜的形式展示出來。如圖2所示,可以看出基因組學、轉錄組學、蛋白質組學、代謝組學是組學共現研究的熱點,文獻數量居于前列。通過連線可以看出,基因組學與轉錄組學的共現文獻量最多,基因組學與蛋白質組學的共現文獻量次之。各類組學之間都存在著錯綜復雜的關系。
研究結果表明,多組學的結合研究已成為組學研究領域的趨勢,整合多組學數據用于藥物重定位和個性化醫療越來越受到重視[15]。因此,相關領域科研人員未來要注意多組學類型的結合研究,從而促進組學研究的進一步發展。
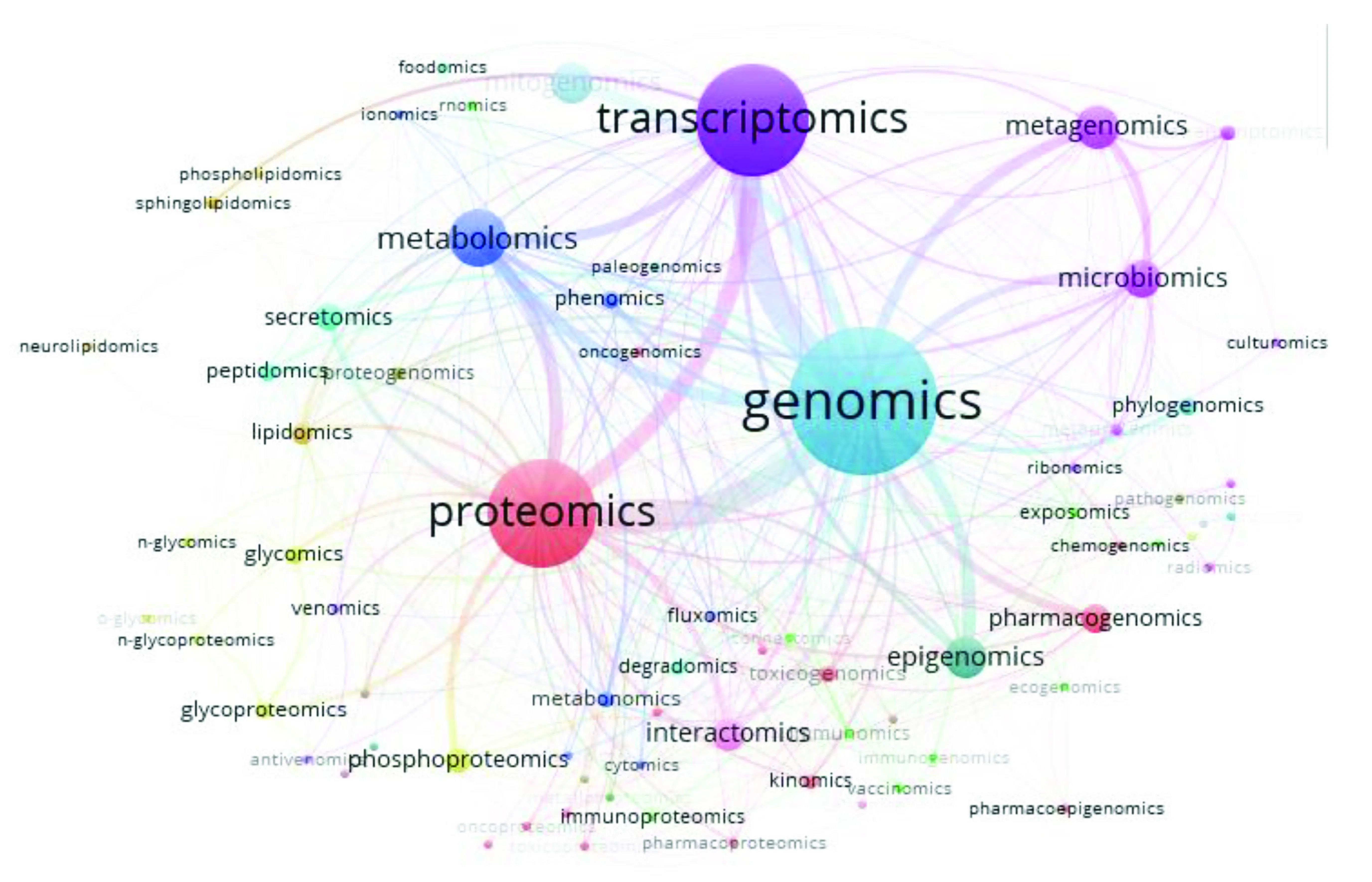
圖2 所有類型組學共現情況
2.4 基因組學脈絡研究
“基因組學”為最早出現的組學類型,且與各類組學都有共現的情況,因此以“基因組學”為主脈絡,展示其余各類組學與“基因組學”共現研究的相關情況,通過主題河圖進行呈現。選取與“基因組學”共現文獻總量排名前15的組學類型,年份從出現多組學共現的第一篇文獻的1995年到2016年進行研究(圖3)。圖3中河流的寬窄代表各類組學與基因組學共現的文獻數的比例,橫坐標為年份的變化。

圖3 “基因組學”與其他各類組學共現論文數變化情況
2.4.1 穩定型增長類型
最早與“基因組學”共現的是“蛋白質組學”。“蛋白質組學”這個概念由Marc Wikins 1994年首次提出[16]。在1995年“基因組學”與其他類型組學共現的5篇論文中,4篇是“基因組學”與“蛋白質組學”的共現,“蛋白質組學”與“基因組學”的共現論文數一直處于穩定增長的趨勢。究其原因,一方面,從分層遞階結構來說,蛋白質系統的粒度較基因組系統粒度粗,蛋白質系統數據處理的復雜度不會超過基因組系統數據處理的復雜度;另一方面,蛋白質的功能性研究距離我們所期望的在細胞水平上研究分子生物學更近,或者說距離在實際應用中所需要的功能研究更近,如在藥物基因組學中的關鍵蛋白質組的尋找[17]。
2.4.2 井噴型增長類型
“轉錄組學”與“基因組學”的共現文獻量隨時間的變化呈井噴式增長,到2016年已成為與“基因組學”共現占比最大的基因類型。究其原因,一是轉錄組學是功能基因組學研究的重要組成部分,是一門在整體水平上研究細胞中所有基因轉錄及轉錄調控規律的學科[18-19];二是隨著新一代高通量基因測序技術運用到轉錄組學研究之中,轉錄組學研究中提供的數據量呈現爆炸式的擴增,拓寬了轉錄組學研究解決科學問題的范圍[14]。
“線粒體基因組學”與“基因組學”的共現論文數量從1995年到2014年一直處于緩慢增長態勢,然而到2015年共現文獻量呈現井噴式增長,成為2016年當年排在“轉錄組學”之后的第二大共現組學類型。究其原因,是由于“線粒體基因組學”在2008年后隨著中國科研人員的加入,半翅目昆蟲線粒體基因組測序進入了迸發階段,在2008-2015年共獲得了89種昆蟲的線粒體基因組,其中81種在中國完成測序。截至2015年5月,美國國立生物技術信息中心共收錄100種半翅目昆蟲的線粒體基因組,其中83個為全線粒體基因組,17個近似完整的線粒體基因組[20]。線粒體基因組的獲取完成在極大程度上推進了線粒體基因組學與基因組學的共同研究。
3 結束語
文獻資料中涵蓋了大量重要信息,能夠從海量的文獻資料中快速挖掘出人們所需求的信息知識,是文獻挖掘技術日益受重視的主要原因。我國“文獻挖掘”多采取在數據庫中檢索所研究的主題對結果進行分析的方式。本文采用獲取PubMed數據庫1896-2016年的全數據的方法,通過對所研究主題的詞根進行識別挖掘,運用社會網絡分析的方法和可視化技術,從組學相關文獻的年度變化趨勢和共現情況方面進行分析,為傳統的文獻挖掘提供了一種新的思路,為學者和研究人員創造了一個知識共享平臺。同時通過分析研究數據,發現后基因組時代的到來把組學研究推向了高潮,無論是數量還是種類都出現了井噴式的增長。多類型組學的融合研究越來越受科研人員的關注,已成為未來組學研究的熱點趨勢。本文的不足在于只從英文文獻著手,研究方法還不夠完備,對多種類型的數據的處理與挖掘還不完善。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
