基于氨基與表面乙烯砜基反應動力學調控配基表面密度
2018-03-29 03:12:13程昉李明洋何煒王漢奇1大連理工大學精細化工國家重點實驗室遼寧大連116023
物理化學學報 2018年2期
程昉,李明洋,何煒,王漢奇1大連理工大學精細化工國家重點實驗室,遼寧 大連 116023
2大連理工大學制藥科學與技術學院,遼寧 大連 116023
3大連理工大學化工學院,遼寧 大連 116023
1 引言
自組裝單分子膜用于表面修飾,能夠在表面引入各種功能性分子,所以被廣泛應用于生物傳感器1,2、酶固定化3、受體-配體相互作用4、蛋白純化5、生物醫學工程6,7等研究工作中。在以上的研究工作中,通常需要對表面生物配基密度進行精準控制,例如在蛋白純化配基密度優化工作中,需要制備不同配基密度的表面從而對最優配基密度進行篩選5;在功能性材料的研究中,也需要對配基表面密度進行優化從而降低非特異性吸附7–9。綜上所述如何實現配基表面密度控制成為了一項具有重要意義的研究工作。
目前應用最為廣泛的表面密度控制方法是混合自組裝,通過調控功能分子與非功能分子的濃度比對表面密度進行調控10–13。例如,Bohmler等14利用混合自組裝的方法得到了一系列密度梯度的Br/CH3和NH2/CH3表面并進行了細菌粘附實驗。Tomohiro等15利用混合自組裝的方法分別在表面修飾了含有 1–3個 6-磺基-N-乙酰基-葡萄糖胺基團的分子并進行了多價態相互作用的研究。Liu等16通過優化混合自組裝比例對固定于表面的蛋白質活性進行優化。本組之前工作17利用混合自組裝對表面糖基功能分子密度進行調控,研究了葡萄糖與Con A蛋白的相互作用。然而,混合自組裝的方法在應用中具有局限性,當進行混合自組裝的兩種分子極性相差較大時會產生相分離從而影響自組裝效果,并且對于不同的自組裝體系不具有普適性。
近年的研究表明利用反應進程控制的方法能夠很好地控制表面密度18,19。反應進程控制法結合表面反應動力學,通過控制配體與表面基團的反應進程實現密度控制。Zhang等19利用Azide-alkyne點擊化學反應實現了表面疊氮基團的密度控制。與混合自組裝的方法相比,操作上更為簡便,可以根據配基的不同調整反應類型,并且對于不同的體系具有普適性。
本組之前工作合成了一種VS (Vinyl Sulfone,乙烯砜基)末端的二硫化合物,利用該化合物構建了VS自組裝表面,并對表面VS基團與巰基、氨基以及羥基在不同 pH條件下的反應動力學進行了研究20。該工作為通過表面反應實現表面密度控制打下了基礎。我們利用自組裝的方法在表面引入VS基團,以ab-NTA為生物配基模型,通過表面催化反應實現NTA密度控制。首先我們利用接觸角的方法對催化 VS與氨基反應的催化劑進行了篩選,并對反應動力學進行了表征。利用反應動力學結果,得到不同配基密度的表面。最后我們利用SPR的方法對不同NTA密度表面與組氨酸標簽蛋白的吸附/解離動態過程進行了研究。
2 實驗部分
2.1 試劑與儀器
Nα,Nα-二(羧甲基)-L-賴氨酸(ab-NTA,97%)購買于Sigma-Aldrich;吡啶二甲酸(97.5%),1-甲基咪唑(99%)購買于 J&K(北京);三苯基膦(分析純)購買于天津市科密歐化學試劑有限公司;HS-C10-COONHS (90%)購買于 ProChimia Surfaces;4-二甲氨基吡啶(DMAP,99%),氯化鎳(分析純),磷酸二氫鈉(分析純),乙二胺四乙酸(EDTA,分析純)購買于阿拉丁生化科技有限公司(上海);咪唑(99%)、鏈霉親和素(SA-6His)購買于索萊寶科技有限公司(北京)。其余有機溶劑均為分析純,購買于阿拉丁生物科技有限公司。
水接觸角測量使用的是接觸角測量儀JC2000D1(中國,上海);表面膜電位表征使用的是NanoBrook Omni (Brookhaven Instruments,美國);表面光電子能譜(XPS)表征使用的是PHI Quantera II Scanning XPS Microprobe (清華大學摩擦學國家重點實驗室提供);SPR實驗均使用的是一臺具有發光二極管光源(λ = 670 nm),高折射率棱鏡(n = 1.61)和雙通道流通單元的Biosuplar-400T SPR光譜儀(Analytical μ-Systems,德國);紫外分光光度計使用的是BioSpectrometer(Eppendorf,德國)。
2.2 自組裝表面制備
2.2.1 金襯底制備
硅片在食人魚溶液(97%濃硫酸和30%過氧化氫比例3 : 1 (V : V)。注意:食人魚溶液對有機物有強烈腐蝕性。)中洗滌30 min后使用大量水和乙醇進行沖洗并用氮氣吹干。利用 Turbo Sputter Coater K575XD (Kent,U.K.)將鈦(5 nm)和金(45 nm)依次蒸鍍到干凈的硅片上。
2.2.2 基于VS的ab-NTA自組裝表面制備
金襯底利用紫外臭氧儀清洗30 min并用乙醇沖洗。隨后,配置 1.0 mmol·L?1的 1,2-二(11-(乙烯砜基)十一烷基)二硫烷溶液(二氯甲烷/乙醇 1 : 1(V : V)),其合成方法之前已有報道20。將清洗干凈的襯底浸入溶液中25 °C放置12 h之后用乙醇沖洗,氮氣吹干。
將 VS自組裝樣品浸入 ab-NTA溶液(10 mmol·L?1ab-NTA、催化劑和 10 mmol·L?1三乙胺溶于 DMSO)中進行反應。之后將樣品浸入EG3-NH2水溶液(10 mmol·L?1,pH 9.5)中反應 12 h進行封閉。最后,用水和乙醇將自組裝樣品沖洗干凈,氮氣吹干,4 °C避光保存。
2.2.3 基于NHS的NTA自組裝表面制備
將清洗干凈的金襯底浸入 1.0 mmol·L?1的HS-C10-COONHS乙醇溶液中25 °C放置12 h,之后用乙醇沖洗,氮氣吹干。NHS自組裝樣品浸入ab-NTA 溶液(10 mmol·L?1,pH 8.5 磷酸鹽緩沖液(PBS))中反應 12 h。最后自組裝樣品用水沖洗干凈,氮氣吹干,4 °C避光保存。
2.3 SPR光譜學
為了測量SA-6His蛋白的特異性吸附量,實驗步驟如下:首先通入10 min 0.1 mol·L?1NiCl2溶液進行螯合,其次通入10 min PBS緩沖液(20 mmol·L?1,pH 7.4)進行洗滌,接下來通入30 min的蛋白溶液,隨后通入10 min 含0.5 mol·L?1咪唑的PBS溶液進行洗脫,再10 min PBS進行洗滌,之后通入5 min含20 mmol·L?1EDTA的PBS溶液對表面進行再生,最后同入10 min PBS溶液對表面進行洗滌。在測量BSA非特異性吸附時,洗脫溶液更換為0.01 mol·L?1NaOH溶液。
2.4 吸附平衡和熱力學
含有不同蛋白質濃度(0.001–0.5 mg·mL?1)的緩沖液通過傳感器表面30 min。達到吸附平衡后表面進行再生,利用紫外分光光度計檢測280 nm處吸光度確定蛋白質濃度。等溫吸附曲線利用Langmuir方程進行擬合:

其中Req和Ceq分別表示平衡吸附時的SPR響應值和平衡蛋白濃度。Rmax表示最大結合量,Kd表示解離常數。
3 結果與討論
3.1 催化劑和催化條件篩選
以ab-NTA為生物活性配基,我們對表面VS基團與ab-NTA反應進行了催化劑篩選。由于氨基與乙烯砜基的反應屬于氮參與的邁克爾加成反應21,本文選擇了五種常用邁克爾加成反應催化劑22(如圖1所示),并采用靜態水接觸角對其催化性能進行了表征。VS表面疏水性較強,隨著反應的進行表面逐漸被 ab-NTA的羧基覆蓋,疏水性逐漸降低,表面靜態水接觸角逐漸降低。
不同催化劑催化2和6 h后的VS表面靜態水接觸角變化值如圖 2所示。由結果可以看出,除三乙胺之外的其余四種催化劑均具有催化該反應的能力,其中吡啶二甲酸的催化效率最高,反應6 h后接觸角降低17.0°,添加三乙胺基本沒有反應,表明在反應體系中添加的三乙胺不會對催化結果產生影響。隨后對吡啶二甲酸的用量進行了優化,在0.1%–20%范圍內選擇了5個濃度進行了試驗,催化2 h后檢測接觸角變化值,結果如圖2b所示,結果顯示催化劑用量在 10%之前催化效率隨催化劑用量增加而逐漸升高,當催化劑用量大于 10%時,催化效率基本保持不變。以上結果表明,通過改變催化劑種類和催化劑的量可以對反應進程進行調控。

Scheme 1 表面修飾示意圖

圖1 催化劑化學結構

圖2 (a)不同催化劑條件下接觸角變化曲線;(b)不同吡啶二甲酸濃度下接觸角變化曲線

圖3 吡啶二甲酸作催化劑條件下的(a)接觸角照片和(b)接觸角變化趨勢
3.2 表面反應動力學測定
利用靜態水接觸角的方法同樣可以對吡啶二甲酸催化ab-NTA和表面VS基團反應的反應動力學進行測定。在此選擇了 10%吡啶二甲酸作為催化劑的條件進行研究。通過測量不同反應時間的表面靜態水接觸角,再利用Cassie公式計算可以得到表面覆蓋度23:

其中 χcarboxyl和 χVS分別表示羧基的表面覆蓋度以及相對于羧基VS基團的表面覆蓋度。
由接觸角照片(如圖 3a)可以明顯看出反應前后表面接觸角變化。圖3b為吡啶二甲酸作催化劑條件下金表面接觸角的變化曲線,結果顯示當表面為VS基團時疏水性較強接觸角為66.1° ± 0.6°,隨著反應的進行,反應24 h后接觸角降為42.9° ±0.5°。經測量利用羧基硫醇自組裝得到的羧基表面接觸角為 θcarboxyl= 37.2° ± 1°。由于每個 NTA 分子含有三個羧基,通過換算可以得到不同反應時間VS基團的表面覆蓋度。將VS基團的表面覆蓋度取自然對數與時間作圖得到圖4,可以看出VS表面覆蓋度的自然對數與時間呈線性相關,這表明這一表面反應可以用一級反應動力學進行描述。反應的速率常數和半衰期可以表示為:

其中χVS和χVS0分別表述VS測量的和最初的表面覆蓋度,t和t0分別表示測量時間和最初時間,kobs為反應速率常數,t1/2表示反應半衰期。
通過計算得到吡啶二甲酸催化表面 VS基團與ab-NTA氨基的反應速率常數為0.0012 min?1,半衰期為569 min。從計算結果看出,該反應較為緩慢,易于通過調控反應時間對配基表面密度進行調控。

圖4 VS (乙烯砜基)基團表面覆蓋度的自然對數
3.3 表面膜電位和XPS表征
為了對修飾前后表面進行表征,分別利用表面膜電位和XPS的方法對VS表面和ab-NTA表面進行了檢測。表面膜電位方法通過對表面電位變化進行檢測,可以反映出表面基團的變化。
如圖 5所示為不同表面的表面膜電位,裸金表面的面電位是?28.33 mV,這是由于金表面含有大量自由電子造成的。當表面修飾上乙烯基砜二硫化化合物后變為?5.45 mV,吡啶二甲酸催化表面VS與ab-NTA反應12 h后,由于表面羧基增多,表面電位降為?15.33 mV。表面膜電位的變化表明了VS二硫化合物的自組裝和ab-NTA與VS基團的反應均成功進行。為進一步對表面結構進行表征,我們對 VS表面和 ab-NTA表面進行了 XPS表征。結果如圖6所示為XPS譜圖,從S 2p譜呈現出兩個峰,分別裂分為峰面積1 : 2的雙峰,其中163 eV周圍的峰歸屬為S-Au峰,169 eV周圍的峰歸屬為砜基峰,這表明VS二硫化合物成功組裝到了金表面。VS表面的C 1s譜在進行擬合后可以分為兩部分:285 eV處的峰歸屬為亞甲基鏈的峰,另一處285.6 eV處的峰歸屬為與砜基相連的碳原子的峰。與VS表面相比ab-NTA表面的C 1s譜在287 eV和288.5 eV處出現兩個峰,分別可以歸屬為與亞胺相連的碳原子的峰和羧基上碳原子的峰。由兩種表面的N 1s譜可以看出,VS表面沒有檢測到N元子,ab-NTA表面在400.4 eV處有N元素的峰,可以歸屬為亞胺的峰。結果表明ab-NTA與表面VS基團成功發生了反應。

圖5 表面膜電位

圖6 (1) VS表面和(2) ab-NTA表面的XPS表征結果
3.4 ab-NTA表面生物功能表征
NTA基團在與鎳離子螯合之后可以與組氨酸上的咪唑殘基發生配位,從而可以特異性結合組氨酸標簽的蛋白24。因此通過SPR對NTA表面的蛋白質結合動力學和熱力學進行研究,可以對表面 NTA密度進行表征。本文選用的蛋白是SA-6His。以NHS活化羧基的方法25,26作為對照。
基于VS和NHS的ab-NTA表面均反應12 h以保證充分反應。如圖7所示為基于VS的ab-NTA表面在通入不同濃度蛋白時的SPR曲線。從圖中可以看出隨著蛋白濃度的升高,其結合量呈現明顯的梯度增高。在通過咪唑洗脫和PBS洗滌之后能夠回到基線,表面基本沒有蛋白質殘留,這表明 SA-6His蛋白與表面的結合為特異性結合。在通過0.25 mg·mL?1蛋白時可以看到蛋白結合量首先迅速增高,隨后出現緩慢降低的現象。我們推測,在通入高濃度蛋白時,蛋白與表面NTA首先形成了單價態結合,隨著蛋白溶液的持續通過,蛋白與NTA逐漸轉變為更穩定的多價態結合,結合量隨之有所降低。如圖8所示,選擇通蛋白30 min時的響應值作為該濃度下的結合量,利用Langmuir方程對數據進行擬合,得到了NTA表面對 SA-6His和 BSA的等溫吸附曲線(圖 8),并與NHS方法進行對比。通過計算可以得到了兩種表面的 Kd和 Rmax(表 1)。
對于 SA-6His蛋白的等溫吸附,基于 VS的NTA 表面 Kd為 0.60 × 10?6mol?1,基于 NHS 的 NTA表面 Kd為 1.01 × 10?6mol?1,由此可以看出基于 VS的NTA表面對于SA-6His蛋白的結合強度更高。從最大結合量來看,基于VS的NTA表面的最大結合量為693,基于NHS的NTA表面的是457。
以上結果表明,基于VS的NTA表面修飾方法較基于NHS的NTA表面修飾方法能夠提供更高的結合強度和結合量,說明相對于NHS的方法基于 VS的表面修飾方法可以提供更高的配基密度。由圖8也可以看出兩種表面對BSA的非特異性吸附均很低,表明基于VS的方法同樣具有較好的抗非特異性吸附能力。
由于NHS酯在堿性水溶液中易水解,導致反應過程伴隨著水解使得基于NHS方法的配基表面密度難以提高。而基于VS的反應符合一級反應動力學,最終可以達到較高的反應效率,所以能夠提高供更高的配基表面密度。與NHS方法相比,基于 VS的方法反應效率更高,配基表面密度更高,并且具有很好抗非特異性吸附能力。

圖7 蛋白質與VS-ab-NTA表面的SPR結合曲線
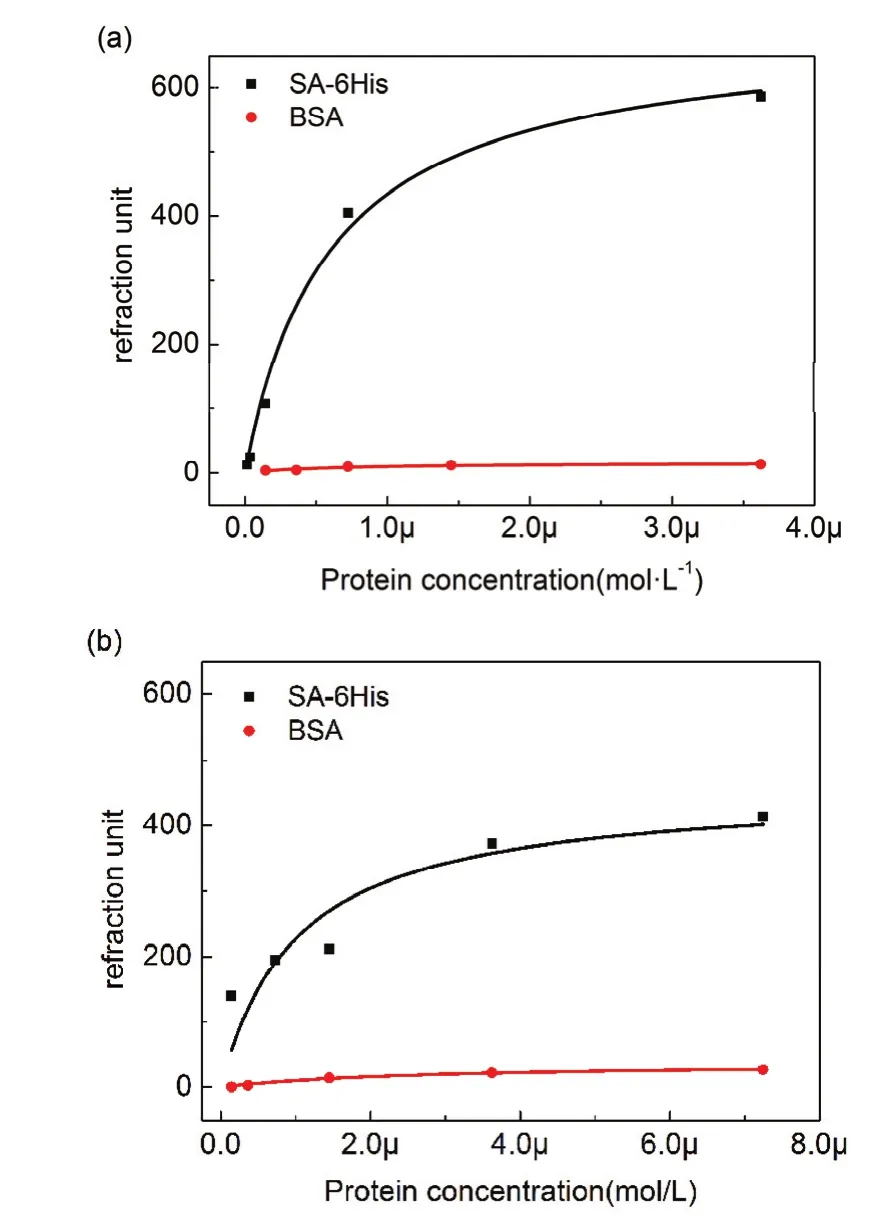
圖8 基于 VS (a)和 NHS (b)的ab-NTA表面的等溫吸附曲線

表1 基于VS和NHS的ab-NTA表面的Kd和RmaxTable 1 Kd and Rmax of ab-NTA surface base on VS and NHS.
3.5 NTA配基密度控制
結合上文催化劑篩選以及反應動力學結果,分別利用吡啶二甲酸催化0.5、2、12 h以及利用三苯基膦催化12 h得到四種不同ab-NTA密度的SPR芯片。利用 SPR對四種芯片分別進行了SA-6His蛋白的靜態吸附實驗。如圖9 (Cat.1)所示為吡啶二甲酸作為催化劑催化不同時間得到的三種不同配基密度的芯片的靜態吸附曲線,從圖中可以看出種不同催化時間芯片的靜態吸附曲線呈現明顯的梯度。圖9中12 h (Cat.2)為三苯基膦作為催化劑催化12 h表面的靜態吸附曲線,與圖9 12 h (Cat.1)曲線對比可以看出,兩個靜態吸附曲線具有明顯梯度。上述結果再次證明通過控制反應時間和催化劑種類能夠對配基表面密度進行調控。
利用上文反應動力學結果可以計算出四種芯片表面羧基的表面覆蓋度分別為14.5%、41.2%、80.6%和34.5%。根據報道羧基硫醇自組裝膜的密度為6.98 molecule nm?227。通過估算可以得出四種表面的ab-NTA密度分別約為0.34、0.96、1.88和 0.80 molecule·nm?2。利用 Langmuir方程對數據進行擬合后可以到的表 2所示結果,四種不同配基密度表面的蛋白最大結合量分別為183、504、693和282,可以看出蛋白最大結合量與配基表面密度呈正相關,隨著配基密度的增加,表面最大結合量增大。
表 2結果顯示四種不同配基密度表面對SA-6His蛋白的 Kd值分別為 1.13 × 10?6、0.62 ×10?6、0.60 × 10?6和 0.58 × 10?6mol?1。可以看出吡啶二甲酸催化0.5 h的表面配基與SA-6His蛋白的結合強度為 1.13 × 10?6mol?1,明顯低于另外三種表面。由表面ab-NTA密度可以估算得到四種表面的NTA分子平均間距分別約1.7、1.0、0.7和1.1 nm。組氨酸標簽與NTA的結合屬于多價態結合28,每兩個咪唑基團可以與一個鎳離子配位,六個組氨酸標簽的長度約為2.0 nm。對比以上數據可以看出,在低密度表面上每個組氨酸標簽蛋白只能與1–2個NTA分子絡合,此時蛋白與NTA之間多數形成單價態結合,所以結合強度較低。吡啶二甲酸催化2、12 h以及三苯基膦催化12 h的表面上每個蛋白則能夠與2–3個NTA分子絡合,可以形成多價態相互作用,結合強度明顯提高。
上述實驗結果通過控制表面 VS基團與氨基的催化反應時間以及催化劑種類實現了配基表面密度調控,并建立了表面密度與性能之間的構效關系。該工作為表面密度控制提供了新的方法。對于生物傳感器修飾、蛋白純化配基密度優化、生物醫學材料表面修飾等研究工作,利用本文的配基表面密度控制方法可以在表面模擬不同配基密度的材料表面環境,結合SPR、QCM等研究方法可以簡便地不同配基密度表面的特異性/非特異性結合性能進行表征,提高了密度篩選的效率。
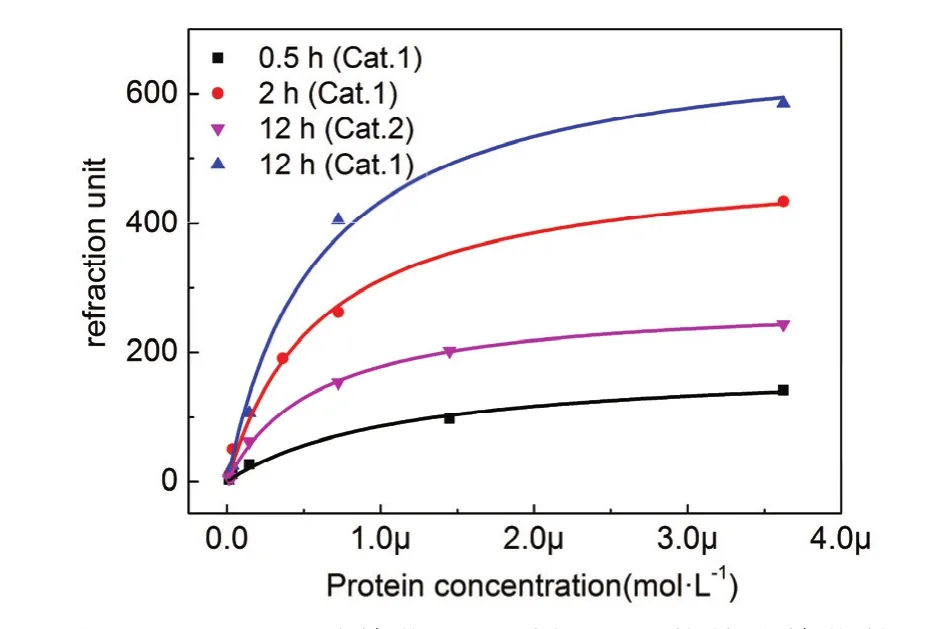
圖9 吡啶二甲酸催化不同時間和三苯基膦催化的表面的等溫吸附曲線

表2 不同反應時間和催化劑催化的ab-NTA表面的Kd和RmaxTable 2 Kd and Rmax of ab-NTA surface with different reaction time and catalysts.
4 結論
本文利用靜態水接觸接觸角的方法對一系列N、P中心的催化劑催化表面VS基團與氨基反應的催化性能進行了表征,結果顯示吡啶二甲酸作為表面VS與氨基反應的催化劑具有最高的催化效率,DMAP、一甲基咪唑和三苯基膦也可以催化該反應。對吡啶二甲酸催化反應動力學的研究結果顯示該反應符合一級反應動力學,其反應速率常數為0.0012 min?1,半衰期為569 min。本文以ab-NTA作為配基,分別利用傳統的NHS活化法和VS法對金表面進行了修飾,并利用SPR技術對表面蛋白靜態吸附進行了研究,結果表明利用VS的方法能夠提供更高的配基表面密度。根據反應動力學結果分別利用不同催化劑對 VS表面進行不同時間的修飾,并結合SPR對不同反應時間的表面進行了蛋白質靜態結合實驗,結果表明通過控制反應時間和催化劑種類均可以實現配基表面密度控制。同時,在靜態結合實驗中觀察到了單價態相互作用和多價態相互作用。反應0.5 h的表面由于配基密度低,配基分子間距大,造成蛋白質與配基的結合傾向于單價態結合,結合強度較高密度表面有明顯的降低。
本文所發展的催化劑催化的表面乙烯砜基與氨基的反應能夠高效地在表面引入含有氨基的分子,并能夠通過控制反應時間和催化劑種類對表面反應進程進行控制,從而實現對配基表面密度的調控,為表面密度控制提供了新方法。與混合自組裝方法相比本方法可控性強、應用廣泛并且具有一定普適性。
(1) Yuan, P. X.; Deng, S. Y.; Yao, C. G.; Wan, Y.; Cosnier, S.; Shan, D.Biosens. Bioelectron. 2017, 89, 319. doi: 10.1016/j.bios.2016.07.031
(2) Cabanas-Danes, J.; Rodrigues, E. D. J. Am. Chem. Soc. 2014, 136,12675. doi: 10.1021/ja505695w
(3) Nakamura, I.; Horikawa, Y.; Makino, A.; Sugiyama, J.; Kimura, S.Biomacromolecules 2011, 12, 785. doi: 10.1021/bm101394j
(4) Schartner, J.; Hoeck, N. Anal. Chem. 2015, 87, 7467.doi: 10.1021/acs.analchem.5b01823
(5) Cheng, F.; Li, M. Y.; Wang, H. Q.; Lin, D. Q.; Qu, J. P. Langmuir 2015, 31, 3422. doi: 10.1021/la5044987
(6) Rowley, J. A.; Mooney, D. J. J. Biomed. Mater. Res. 2002, 60, 217.doi: 10.1002/jbm.1287
(7) Shoffstall, A. J.; Everhart, L. M. Biomacromolecules 2013, 14, 2790.doi: 10.1021/bm400619v
(8) Chen, X. W.; Pei, D. H. J. Comb. Chem. 2009, 11, 604.doi: 10.1021/cc9000168
(9) Shao, Q.; Jiang, S. Y. J. Phys. Chem. B 2014, 118, 7630.doi: 10.1021/jp5027114
(10) Tomohiro, H.; Kenji, W. J. Phys. Chem. C 2009, 113, 18795.doi: 10.1021/jp906494u
(11) Subramanian, A.; Irudayaraj, J.; Ryan, T. Sensor. Actuat. B:Chem. 2006, 114, 192. doi: 10.1016/j.snb.2005.04.030
(12) Ma, H.; Wells, M.; Beebe, T. P. Jr.; Chilkoti, A. Adv. Funct.Mater. 2006, 16, 640. doi: 10.1002/adfm.200500426
(13) Bain, C. D.; Whitesides, G. M. J. Am. Chem. Soc. 1988, 110,6560.
(14) Bohmler, J.; Ponche, A.; Anselme, K.; Ploux, L. ACS. Appl.Mater. Inter. 2013, 5, 10478. doi: 10.1021/am401976g
(15) Tomohiro, F.; Yoshiko, M. Bioconjugate Chem. 2010, 21,1079. doi: 10.1021/bc100053x
(16) Liu, Y. T.; Yan, L.; Sun, L. M.; Li, H. Q.; Li, H. H. Chem. Eng.(China) 2014, 42, 69. [劉玉婷, 顏莉, 孫立民, 李慧琴, 李海華. 化學工程, 2014, 42, 69.]doi: 10.3969/j.issn.1005-9954.2014.03.014
(17) Cheng, F.; Wang, H. Q.; Xu, K.; He, W. Acta Phys. -Chim.Sin. 2017, 33, 426. [程昉, 王漢奇, 許曠, 何煒. 物理化學學報, 2017, 33, 426.] doi: 0.3866/PKU.WHXB201609291
(18) Eugene W. L.; Chan, M. N. Y. J. Am. Chem. Soc. 2006, 128,15542. doi: 10.1021/ja065828l
(19) Zhang, S.; Maidenberg, Y.; Luo, K.; Koberstein, J. T.Langmuir 2014, 30, 6071. doi: 10.1021/la501233w
(20) Wang, H. Q.; Cheng, F.; Li, M. Y.; Peng, W.; Qu, J. P.Langmuir 2015, 31, 3413. doi: 10.1021/la504087a
(21) Esteves, A. P.; Silva, M. E.; Rodrigues, L.M.;Oliveira-Campos, A. M. F.; Hrdina, R. Tetrahedron Lett. 2007,48, 9040. doi: 10.1016/j.tetlet.2007.10.077
(22) Wang, C.; Qi, C. Z. Tetrahedron 2013, 69, 5348.doi: 10.1016/j.tet.2013.04.123
(23) Cassie, A. B. D.; Baxter, S. Trans. Faraday Soc. 1944, 40,546.
(24) Kim, E. J.; Chung, B. H.; Lee, H. J. Anal. Chem. 2012, 84,10091. doi: 10.1021/ac302584d
(25) Maalouli, N.; Gouget-Laemmel, A. C. Langmuir 2011, 27,5498. doi: 10.1021/la2005437
(26) Pei, J.; Tang, Y.; Xu, N.; Lu, W.; Xiao, S. J.; Liu, J. N. Sci.China Chem. 2010, 54, 526. doi: 10.1007/s11426-010-4128-3
(27) Shin-ichiro, I.; Takashi, K. J. Electroanal. Chem. 1997, 428,33. doi: 10.1016/S0022-0728(97)00006-5
(28) Suman L.; Jacob, P. J. Am. Chem. Soc. 2005, 127, 10205.doi: 10.1021/ja050690c
猜你喜歡
兒童故事畫報(2019年5期)2019-05-26 14:26:14
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
Coco薇(2016年2期)2016-03-22 02:42:52
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
