AZD9291與STAT3抑制劑對肺癌細胞H1975的協同效應及機制
2018-04-23 08:20:29湯繼春杜瀛瀛潘躍銀
安徽醫科大學學報 2018年3期
湯繼春,袁 媛,金 偉,杜瀛瀛,潘躍銀
目前80%肺癌患者為非小細胞肺癌(non-small cell lung cancer, NSCLC),5年生存率約15%,預后不佳[1]。分子靶向治療在肺癌治療中占有重要地位,第一、二代表皮生長因子受體酪氨酸激酶抑制劑(epidermal growth factor receptor -tyrosine kinase inhibitors,EGFR-TKI)前期治療效果顯著,但半年至一年后出現耐藥[2]。現已發現多種耐藥機制,包括表皮生長因子受體(epidermal growth factor receptor,EGFR)擴增、遺傳損傷、并行旁路的激活等[3]。第三代EGFR-TKI 中AZD9291(osimertinib, 奧斯替尼)已被批準用于T790M陽性NSCLC患者,但耐藥仍不可避免[4]。信號傳導及轉錄激活因子3(signal transducers and activators of transcription, STAT3)在多種腫瘤,如頭頸、肺和乳腺癌中被激活。STAT3的持久活化具有致癌性,有報道[5-6]封阻STAT3信號通路可抑制癌細胞生長,增強抗癌藥物的敏感性。該研究主要檢測AZD9291作用H1975后胞內信號通路變化,以及抑制STAT3通路是否增強AZD9291的抑瘤作用,為以STAT3為靶點的NSCLC治療提供理論和實驗依據。
1 材料與方法
1.1實驗材料RPMI-1640、FBS購自美國Hyclone公司;MTT購自美國Sigma-Aldrich公司;Western blot化學發光劑購自美國Millipore 公司;H1975細胞由廣東省肺癌研究所惠贈;AZD9291由英國Astra Zeneca公司惠贈; STAT3抑制劑(商品名S3I-201)購自美國Santa Cruz Biotechnology公司;Annexin V-FITC凋亡試劑盒購自長沙貝博生物科技公司;磷酸化蛋白激酶B(phosphorylated protein kinase B, p-AKT)和蛋白激酶B(protein kinase B,AKT )抗體、磷酸化細胞外信號調節激酶(phosphorylated extracellular signal-regulated kinase, p-ERK)和細胞外信號調節激酶抗體(extracellular signal-regulated kinase,ERK)、p-STAT3和STAT3抗體均購自美國Cell Signaling Technology公司。
1.2實驗方法
1.2.1細胞培養 H1975細胞培養于含10% FBS、青鏈霉素各100 U/ml的10 ml RPMI-1640培養液中,常規培養在恒溫培養箱,傳代時間為2~3 d,取對數生長期細胞用于研究。
1.2.2MTT法 取96孔板,將H1975細胞接種入內,每孔數量為2 000個,培養箱培養24 h。分為AZD9291單藥組(AZD9291)、STAT3抑制劑S31-201單藥組(S31-201)。單藥實驗組濃度梯度設定為6組,每組6個復孔,使用AZD9291單藥的處理組將濃度設定為為2.625、5.25、10.5、21、42、84 nmol/L;STAT3抑制劑S31-201單藥組實驗濃度分別設計為11.25、22.5、45、90、180、360 μmol/L。后以6種不同濃度AZD9291和STAT3抑制劑聯合給藥培養72 h后,各孔加MTT溶液20 μl,反應4 h后棄去上清液,每孔150 μl DMSO,酶標儀震蕩后在490 nm波長下測量吸光度(optical density,OD)值。細胞的增殖抑制率(%)=[1-(實驗組OD值-空白組OD值)/(對照組OD值-空白組OD值)]×100%。分別計算AZD9291和STAT3抑制劑的半數抑制濃度(half inhibitory concentration,IC50)和聯合指數(combination index,CI)。對于后續的細胞凋亡實驗、Western blot實驗均采用 AZD9291濃度20 nmol/L, STAT3抑制劑S31-201濃度100 μmol/L作為實驗組用藥濃度。
1.2.3流式細胞術檢測細胞凋亡 將H1975接種在6孔板中,密度為1×108個/L ,實驗分組為對照組,AZD9291單藥組,STAT3抑制劑單藥組,以及AZD 9291和STAT3抑制劑聯合用藥組。加入預定濃度的藥物,使其在新培養基中培養72 h。PBS沖洗2遍,胰酶消化收集。離心機離心5 min,棄上清液,保留細胞。分別每管加入Annexin-V結合液400 μl,5 μl的Annexin-V染色,10 μl的PI染色,避光孵育5 min。1 h內置流式機檢測。
1.2.4Western blot法 按1.2.3方法進行分組及藥物處理72 h,消化收集細胞,蛋白裂解液提取蛋白,BCA法測定蛋白濃度,沸水水浴15 min,離心機調為12 000 r/min,高速離心10 min。將樣品加入10% SDS-PAGE中電泳,4 ℃條件下80 V轉膜30 min,后調為120 V轉膜70 min,完成后5%脫脂奶粉室溫下封閉2 h。加入以1 ∶1 000比例稀釋的抗體,4 ℃冰箱過夜。TBST洗滌3遍后,加入二抗(1 ∶5 000),室溫孵1 h,TBST再洗滌3遍。后加入ECL發光劑,暗室中進行顯影結果。各蛋白的灰度值采用Image J軟件分析。
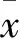
2 結果
2.1AZD9291和STAT3抑制劑對H1975細胞的增殖抑制作用根據前期預實驗結果,選擇藥物作用72 h,MTT結果顯示,AZD 9291和STAT3抑制劑對H1975細胞的IC50值分別為(20.00±3.65)nmol/L和(96.00±15.30)μmol/L,并且隨著藥物濃度的遞增,增殖抑制作用也隨之增強,具有濃度依賴性(F=113.89、138.68,P<0.05)。見圖1。
不同濃度的AZD 9291聯合STAT3抑制劑處理H1975細胞72 h,相對于單藥組,兩藥聯合后增殖抑制作用明顯增強(F=36.39,P<0.05),各聯合藥物劑量組的CI均<1,表現出協同效應(圖2)。

圖1 MTT法檢測AZD 9291或STAT3

圖2 AZD 9291聯合STAT3抑制劑作用于H1975細胞的CI
2.2AZD9291和STAT3抑制劑聯合促進H1975的凋亡流式細胞儀檢測結果顯示對照組中細胞凋亡率為(3.59±0.62)%,AZD9291和STAT3抑制劑單藥處理組的細胞凋亡率分別為(6.09±0.76)%和(10.13±1.57)%,兩藥聯合后凋亡率上升為(22.18±3.24)%。聯合用藥組的細胞凋亡率較單獨用藥組均明顯增高,差異有統計學意義(F=84.043,P<0.05)。見圖3。
2.3AZD9291和STAT3抑制劑單藥及聯合用藥對H1975細胞AKT、p-AKT、ERK、p-ERK、STAT3、p-STAT3蛋白表達的影響如圖4示,檢測顯示與對照組相比較,AZD9291可明顯上調p-STAT3 蛋白的水平,而p-AKT和p-ERK蛋白的表達水平明顯下降,差異有統計學意義(F=45.80、20.66、232.02,P<0.05);兩藥聯合組p-STAT3蛋白水平與AZD9291單藥組相比明顯下降,差異有統計學意義(F=29.80,P<0.05),見圖5。
3 討論
STAT蛋白家族可以被多種細胞因子受體激活,其中最受關注的為STAT3,多種促炎因子和生長因子可以使其活化,在多種人類惡性腫瘤中STAT3出現明顯異常激活,其參與了多種腫瘤的致癌信號通路的傳遞及胞內信號轉導通路的信號傳遞[7]。在正常情況下,胞內STAT3信號的激活訊速但持續時間短暫,受多種負性調控因子的調節。近年多種研究[8-10]證實,在白血病、肺癌、肝癌、乳腺癌及前列腺癌等中STAT3均有異常而持續的激活。STAT3 參與了腫瘤細胞的增殖、分化、侵襲、轉移、血管生成及抗凋亡作用。抗凋亡蛋白、細胞周期蛋白、基質金屬蛋白酶和血管內皮生長因子等的表達增高均與STAT3表達存在密切關系[7]。因此,深入研究STAT3 在腫瘤中機制,從而設計針對STAT3 靶向治療藥物,可能為腫瘤治療的提供一個有效途徑。
以表皮生長因子受體為靶點的分子靶向治療在 NSCLC 的治療中日益占據重要位置,然而患者對靶向藥物有耐藥現象,其中大約一半耐藥機制是 EGFR 的T790M 位點突變或c-MET 擴增[11-12],但是余下耐藥機制尚未完全闡明,部分研究[8-10]表明與STAT3的異常活化有關。研究[13-14]表明STAT3 可以增加腫瘤細胞在治療藥物中的適應能力,導致腫瘤產生耐藥。Chiu et al[13]研究發現p53的穩定劑 RITA 與吉非替尼聯合增加對 H1650的敏感性,主要機制為抑制STAT3的活性。同樣的,STAT3 抑制劑WP1066 或小干擾RNA可以導致H1650 的增殖減低;以上證實STAT3對維持H1650 在吉非替尼中的耐藥有至關重要的作用。Kim et al[14]研究表明,選擇H1975 和 PC9/GR這兩種攜帶T790M 突變的 NSCLC 細胞株進行檢測,發現阿法替尼可以激活 IL-6R/JAK1/STAT3 信號通路,主要通過自分泌IL-6因子。使用信號通路阻斷劑如JAK 抑制劑或是IL-6中和抗體可以顯著增強了阿法替尼的抗腫瘤的敏感性。以上結果同樣在小鼠體內移植瘤實驗中證實。
有研究[15]顯示通過一些藥物提前處理后能夠直接或是間接激活STAT3,使得腫瘤出現耐藥。Li et al[15]報道稱在NSCLC中,厄洛替尼抑制PTPMeg2 表達,使得STAT3中Y705磷酸化并上調bcl-2/bcl-xl的基因和蛋白表達,存活信號通路的激活使得細胞對厄洛替尼耐藥。厄洛替尼聯合STAT3 抑制劑 Nidosamid或是小干擾RNA可以逆轉HCC827的耐藥,以上結果同樣在移植瘤中證實,厄洛替尼聯合STAT3 抑制劑增強瘤組織的縮小和凋亡。這些發現揭示了新的EGFR-TKI可能的耐藥機制,為克服肺癌靶向藥物耐藥提供了一個新的治療策略。
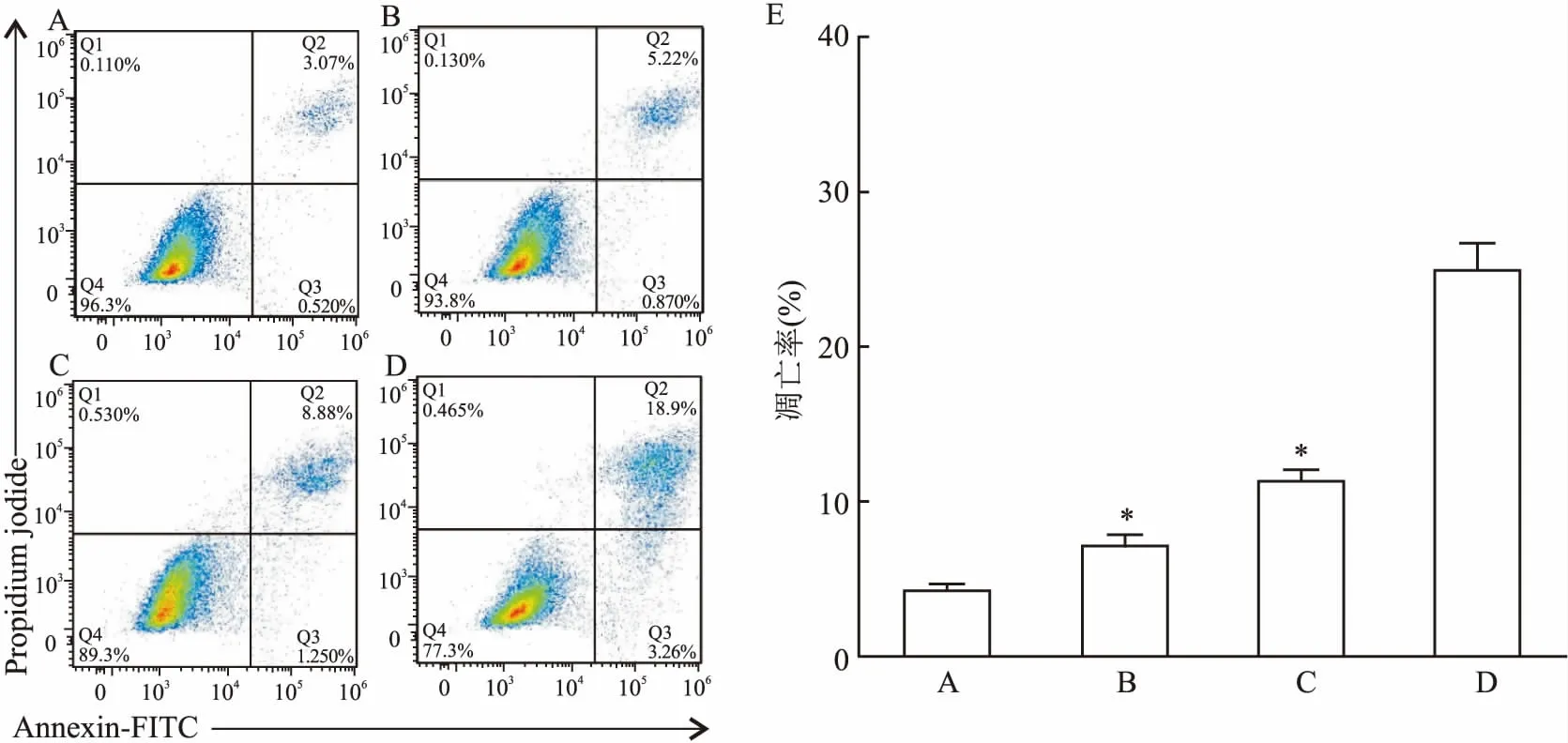
圖3 流式細胞術法檢測AZD 9291和STAT3抑制劑單藥及聯合作用對H1975細胞凋亡率的影響
A:對照組;B:AZD9291單藥組;C:STAT3抑制劑單藥組;D: AZD9291+STAT3抑制劑聯合用藥組;E:AZD9291與STAT3抑制劑單藥及聯合作用對H1975細胞凋亡率影響的比值;與AZD9291+STAT3抑制劑聯合用藥比較:*P<0.05
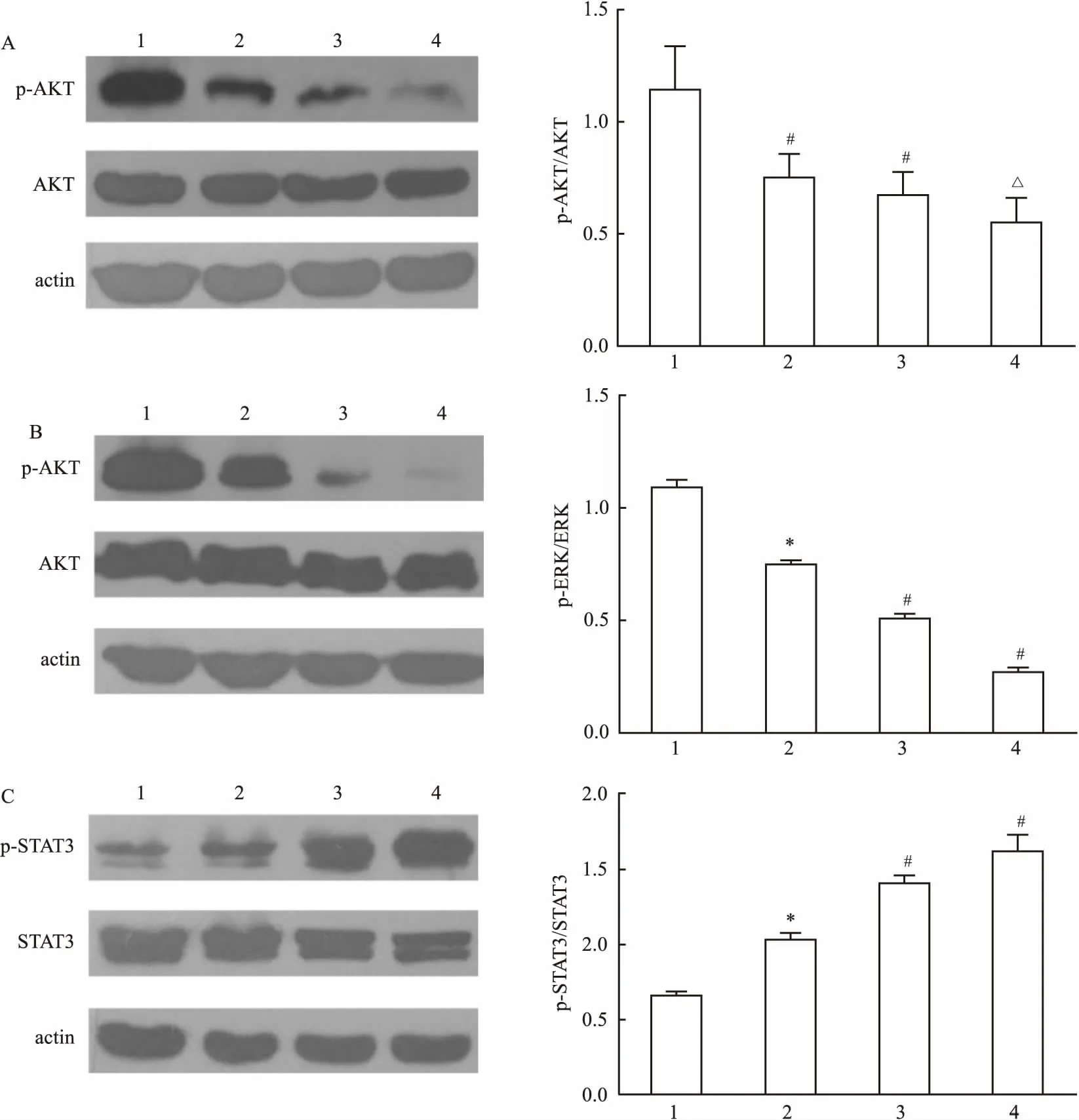
圖4 AZD9291作用于H1975細胞后信號通路蛋白的變化
A:AZD9291單藥作用細胞后p-AKT及AKT蛋白變化;B:AZD9291單藥作用細胞后p-ERK及ERK蛋白變化;C: AZD9291單藥作用細胞后p-STAT3及STAT3蛋白變化;1:對照組;2:10 nmol/L;3:20 nmol/L;4:40 nmol/L;與對照組比較:*P<0.05;與10 nmol/L組比較:#P<0.05;與20 nmol/L組比較:△P<0.05
圖5 AZD 9291和STAT3抑制劑單藥及聯合后對H1975信號通路中p-STAT3蛋白表達的影響
為了解使用AZD9291后細胞內主要細胞通路變化情況,本實驗主要檢測了細胞內EGFR下游主要三條信號通路的變化。其中AKT及ERK通路在低劑量組即出現明顯抑制,但STAT3信號通路卻異常激活,且磷酸化STAT3隨劑量增高而增高。雖然AZD9291治療效果顯著,但不可避免地會出現耐藥,STAT3的異常激活可能為AZD9291的治療效果欠佳的一個因素,所以予以AZD9291聯合STAT3抑制劑應用了解其對H1975細胞系增殖情況影響。通過體外實驗,在H1975細胞株中顯示AZD 9291和STAT3抑制劑聯合應用對 H1975 細胞的抗增殖作用明顯強于各單藥組,經過計算其差異有統計學意義,同時,計算CI顯示,AZD9291和STAT3抑制劑聯合的CI<1,表明兩者聯合具有明顯的協同作用。同時,通過流式細胞儀法對凋亡進行檢測,本研究顯示AZD9291和STAT3抑制劑聯合可明顯促進 H1975細胞凋亡的作用。AZD9291在作用H1975后出現STAT3活性明顯增高,其可能進一步激活了下游信號通路抗凋亡蛋白,導致細胞對抑制劑敏感性降低,所以聯合應用STAT3抑制劑后AZD9291其抑制細胞增殖作用增強。
綜上所述,AZD9291和STAT3抑制劑聯用對攜帶T790M突變的H1975細胞株具有良好的協同作用,可增強細胞對AZD9291的敏感性,為臨床EGFR-TKIs 獲得性耐藥患者的治療提供新的實驗室依據及治療策略。但本實驗僅為體外細胞研究,尚需進一步的動物實驗及臨床試驗來驗證其在體內的有效性及安全性。
[1] Siegel R L, Miller K D, Jemal A.Cancer statistics,2015 [J].CA Cancer J Clin,2015,65(1):5-29.
[2] 萬宜濤,袁 媛,潘躍銀,等.酪氨酸激酶抑制劑高劑量脈沖式給藥克服非小細胞肺癌獲得性耐藥的體外觀察研究[J]. 安徽醫科大學學報, 2016, 51(4):516-20.
[3] Niu F Y, Wu Y L. Novel agents and strategies for overcoming EGFR TKIs resistance [J]. Exp Hematol Oncol, 2014, 3(1):2.
[4] Govindan R.Overcoming resistance to targeted therapy for lung cancer [J]. N Engl J Med,2015,372(18) :1760-1.
[5] Geiger J L, Grandis J R, Bauman J E. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations [J]. Oral Oncol, 2016, 56:84-92.
[6] Zhao C, Li H, Lin H J, et al. Feedback activation of STAT3 as a cancer drug-resistance mechanism [J]. Trends Pharmacol Sci, 2016, 37(1):47-61.
[7] Siveen K S, Sikka S, Surana R, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors [J].Biochim Biophys Acta,2014,1845(2):136-54.
[8] Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma [J]. Cancer Cell, 2015,27(4):516-32.
[9] Wen W, Wu J, Liu L, et al. Synergistic anti-tumor effect of combined inhibition of EGFR and JAK/STAT3 pathways in human ovarian cancer [J]. Mol Cancer, 2015, 14:100.
[10] Deshmukh S K, Srivastava S K, Bhardwaj A, et al. Resistin and interleukin-6 exhibit racially-disparate expression in breast cancer patients, display molecular association and promote growth and aggressiveness of tumor cells through STAT3 activation[J]. Oncotarget, 2015, 6(13):11231-41.
[11] Wu S G, Liu Y N, Tsai M F, et al. The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients [J]. Oncotarget, 2016, 7(11):12404-13.
[12] Chabon J J, Simmons A D, Lovejoy A F, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients[J]. Nat Commun, 2016,7:11815.
[13] Chiu H C, Chou D L, Huang C T, et al. Suppression of Stat3 activity sensitizes gefitinib-resistant non small cell lung cancer cells [J]. Biochem Pharmacol,2011,81(11): 1263-70.
[14] Kim S M, Kwon O J, Hong Y K, et al. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation [J]. Mol Cancer Ther,2012, 11(10): 2254-64.
[15] Li R,Hu Z,Sun S Y,et al.Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non-small cell lung cancer [J]. Mol Cancer Ther,2013,12(10):2200-12.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
保健醫苑(2022年5期)2022-06-10 07:46:38
昆明醫科大學學報(2022年1期)2022-02-28 07:43:40
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
電子制作(2018年11期)2018-08-04 03:25:42
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
