基于RAD重測序技術開發煙草品種SNP位點
2018-05-14 09:36:57任民程立銳劉旦蔣彩虹楊愛國
中國煙草科學 2018年3期
關鍵詞:煙草
任民 程立銳 劉旦 蔣彩虹 楊愛國

摘要:利用限制性內切酶位點標簽(RAD)技術,通過對10份供試煙草材料的基因組簡化重測序,發掘了煙草高通量SNP位點,為煙草基因組學提供標記信息。結果表明,本研究共獲得了44.33 Gb的Clean data數據,平均覆蓋度1.01 X,共鑒定到291 770個SNP位點,SNP位點間的平均間距為10.066±29.801 kb。發掘到的SNP位點能夠覆蓋整個基因組,但在不同染色體部位上的分布密度存在一定差異,在17號染色上半臂的存在一段大范圍的SNP密集區域。SNP變異類型以轉換為主,通過功能注釋在基因區域發現45 049處SNP位點。利用SNP分型信息,計算了供試品種間的遺傳距離,平均為0.29,臺煙8號的遺傳背景與其他品種相對最遠。該結果將為煙草QTL定位、候選基因發掘、親本組配等研究提供科研依據。
關鍵詞:煙草:限制性內切酶位點標簽:重測序:單核苷酸多態性
煙草品種是煙葉生產的基礎,在行業可持續發展與產業升級中發揮著重要的作用。而且品種本身也是一類重要的種質資源,其研究價值不僅在于所攜帶的優良基因或等位變異,也包括這些優良基因的組合方式、組成特點,及其所承載的育種經驗和成果。從育種技術發展的趨勢來看,對品種的綜合性狀提升提出了越來越高的要求。深入發掘種質資源、多途徑創新種質、擴大遺傳背景等研究已經越來越受到重視,分子育種、基因組學等新技術新理論已經成為育種技術的研究熱點。近年來,圍繞煙草育種骨干親本、種質資源,開展了大量的分子標記研究。現已構建了煙草高密度SSR遺傳圖譜,并開展了重要性狀的QTL定位和GWAS分析等研究。但煙草是異源四倍體(2n=4X=48),基因組龐大結構復雜,且品種間遺傳多樣性較低。因此隨著研究的深入,以SSR為代表的二代分子標記技術,在標記密度、數據通量和研究效率等方面的限制都制約著進一步的應用。近年來高通量測序技術取得了快速發展,限制性內切酶位點標簽(Restriction-Site Associated DNA,RAD)成為當前簡化基因組測序策略中運用較為廣泛的測序技術,該技術與海量平行測序技術偶聯可實現極高的分析效率,且成本相對較低。能夠廣泛的運用于全基因組關聯分析,高密度遺傳連鎖圖譜的構建,目標性狀調控相關基因組區段或候選基因快速定位,個體間遺傳多態性分析等。為此本研究擬利用高通量測序技術(RAD)對10份常用的煙草病毒病(TMV、CMV)抗感鑒定品種進行重測序,發掘單核苷酸多態性(single NucleotidePolymorphism,SNP)位點,深入了解供試品種的遺傳多樣性。對于促進優異基因資源發掘、抗病品種分子育種等有重要的研究和實踐意義。
1材料與方法
1.1供試材料
本研究供試品種均為普通煙草(Nicotianatabacum L)種烤煙類型,由國家煙草種質資源中期庫提供,其品種名稱及編號見表1。可通過種質資源編號在中國煙草種質資源網(http://www.ycsjk.com.cn/)檢索供試品種的資源調查信息。
1.2全基因組DNA提取
供試品種播種后培養至苗期,取幼葉組織采用稍加改良的SLS法提取全基因組DNA。(1)將磨好的葉片放入2 mLEP管中,加SLS提取液800μL,搖晃5 min至搖勻:(2)加入等體積的酚氯仿異戊醇混合液(V:V:V=25:24:1),搖晃5 min至搖勻,然后12000 rpm離心10 min:(3)吸取上清600μL至一新的1.5 mL離心管,加等體積預冷的異丙醇(-20℃)沉淀DNA:(4)12000rpm離心10min,棄上清。用75%乙醇洗滌1次,無水乙醇再漂洗1次:(5)置于超凈臺內晾30~60min至完全干燥無酒精殘留后,加100~200μLddH20溶解。最后用1.0%的瓊脂糖凝膠電泳和NanoDrop 2000分光光度計對DNA質量進行檢測。
1.3參考基因組
本研究SNP鑒定和功能預測采用的參考基因組為普通煙草栽培品種紅花大金元的全基因組組裝序列(第2版),其基因組序列數據和基因注釋信息見中國煙草基因研究中心煙草基因組數據庫(http://218.28.140.17/)。參考基因組序列總長4411.73 Mb,其中組裝到24條染色體的序列總長2939.14Mb,此外還有29 802條Scaffold序列,其總長為1472.58 Mb。
1.4簡化基因組(RAD)測序及SNP鑒定
RAD簡化基因組測序及SNP鑒定由華大基因有限公司完成,采用Eeor I限制性內切酶進行酶切隨機打斷基因組DNA,測序儀器為Illumina Hiseq2000:從測序后CleanData數據中鑒定SNP采用了GATK-3.2-2流程(https://www.broadinstitute.org/gatk/)。
1.5數據分析
采用DnaSP 6.01281計算供試群體的核苷酸多態性(Nucleotide Polymorphism)兀值和每位點核苷酸多態性指數[Tbeta(per site)from Etal 0/b0值:采用MEGA 7.0.9軟件根據Maximum CompositeLikelihood模型計算品種間兩兩遺傳距離:采用SnpEff4.1g軟件對鑒定到的SNP進行功能注釋.采用Python 2.7.2計算機語言和P1L(Python ImageLibrary)圖像處理函數庫,按照滑動窗口(slidingwindow)方法統計SNP位置信息并繪制分布密度熱圖,窗口長度(window length)為1.0 Mb,步長(step size)為0.5 Mb:其他數據統計和圖表繪制采用EXCEL 2013完成。
1.6數據獲取
本研究的測序Clean data序列數據,SNP群體分型信息均已提交至中國煙草基因研究中心煙草基因組數據庫(http://218.28.140.17/),數據庫用戶在使用GBrowser瀏覽普通煙草栽培種紅花大金元基因組數據時,可通過添加SNP信息軌道(Track)訪問本研究的相關數據。
2結果
2.1供試品種的基因組重測序
采用RAD測序技術對10份供試品種進行了基因組重測序,獲得原始堿基序列片段(reads)后,又經質量控制和數據過濾,生成高質量的CleanData作為本研究的分析基礎。由表2看出,在供試品種上,測序得到的總堿基數從最少3 016.48 Mb(L9)到最多10032.99Mb(L1),平均為4432.52Mb:按普通煙草基因組大小約為4.4 Gb計算,基因組覆蓋度從供試品種L9的0.69 x到供試品種L1的2.28 X,平均覆蓋度1.01 X:供試品種的平均GC含量和平均Q20比例分別為38.66%和97.65%,其變異系數(c功分別僅為0.31%和0.30%,表明GC含量和Q20在品種間的離散程度低。
2.2供試品種的SNP位點發掘
完成測序后,按照GATK流程開展了供試品種的SNP位點鑒定研究。由表3看出,在參考基因組染色體范圍內,共發掘到291770個SNP位點。SNP數量最多的染色體為17號,共鑒定到33 807處,最少的為24號,僅4527處。SNP位點間的平均間距為10.066±29.801 kb,不同染色體的SNP平均間距亦不相同,間距最小的染色體為17號(平均間距為2.58±16.52 kb),間距最大的染色體為10號(平均間距15.32±39.34 kb)。將24條染色體的SNP密度分布繪制成熱圖(圖1),可發現本研究發掘到的SNP位點能夠覆蓋全部染色體的各個區段,但SNP位點在染色體上的分布密度由圖1可知存在明顯的差異,在2號、13號、17號等染色體上存在較大范圍的SNP高密度區域,尤其以17號染色體上半臂的SNP密度最高。
2.3SNP位點的分類與注釋
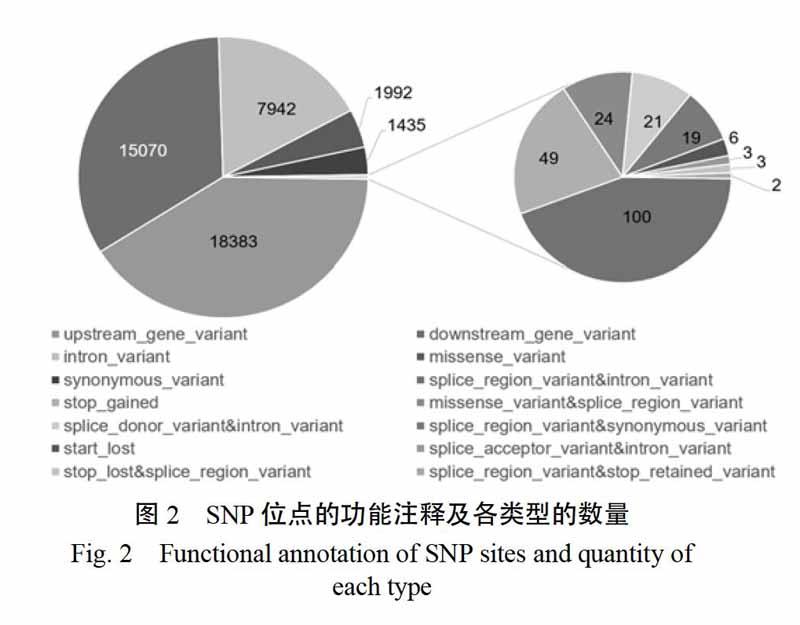
對本研究發掘到的291770個SNP位點進行了分類和注釋。SNP的變異類型中屬于轉換(transitions)的位點有179751處(占63.50%),屬于顛換(transversions)的位點有103329處(占36.50%),轉換與顛換比值為1.74,另外還檢測到8690處非二態性分型的位點(占SNP位點總數的2.98%)。進而結合參考基因組的基因注釋信息,對SNP位點功能進行了注釋,共分成15類。數量最多的一類為基因間區域(intergenic region)SNP,共246721處,占SNP總數的84.56%。其余14類共計45049處SNP位點位于基因區域(含基因上下游3kb以內),基因區域的SNP功能注釋如圖2所示。其中導致氨基酸改變的錯義突變(missensevariant)1992處,翻譯提前終止的突變(stop gained)49處,翻譯無法起始的突變(start lost)6處。
2.4群體遺傳多樣性分析
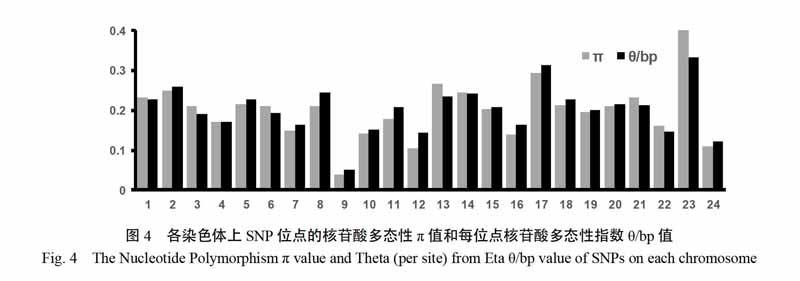
利用供試SNP位點在10份材料上的堿基分型信息,開展了供試群體的遺傳多樣性分析。計算了品種間的遺傳距離,全部供試品種間的平均遺傳距離為0.29,通過圖3可發現,品種L1的遺傳背景與其他品種相對較遠,遺傳距離從0.09到0.46,平均為0.35,而品種L7和L10間的遺傳距離僅為0.03,表明兩份種質的遺傳背景非常接近。在全部供試群體中SNP位點的核苷酸多態性兀值為0.223±0.028,O/bp值為0.221。不同染色體間的遺傳多態性存在明顯的差異(圖4),9號染色體的遺傳多態性程度最低,其兀值為0.040,O/bp為0.05l。23號染色體的多態性最高,其兀值為0.404,O/bp為0.332。
3討論
本研究利用RAD技術對供試品種進行了重測序,獲得了44.33 Gb的Cleandata數據,共鑒定到291 770個SNP位點,SNP位點間的平均間距為10.066±29.801 Kb。分析不同染色體上SNP的分布特點,可見17號染色體的上半臂是一段SNP位點密集的區域,其具體的成因還有待深入研究。在其他煙草基因組研究中也發現了類似的現象,茄科基因組網站(sol Genomics Network,SGN)公布的“HMtabacum 30k Infinium HD consensus map 2015”。(https://solgenomics.net/cview/map.pl?map_versionid=178)煙草高密度SNP遺傳圖譜中報道了一條編號為“Chromosome 17”的連鎖群,該連鎖群的長度較短但SNP數量卻較其他連鎖群多出3~15倍,雖然該連鎖群的編號與本研究所用參考基因組的編號間并無聯系,但其顯著的SNP密度特點與本研究的17號染色體非常一致,故推測很可能是同一條染色體。EDWARDS等對普通煙草的24條染色體的起源進行了分析,發現除“Chromosome Nt17”(編號與SGN網站一致)外其他染色體都能良好的區分為“S基因組起源”和“T基因組起源”,而“Chromosome Nt17”在普通煙草的兩個祖先種林煙草和絨毛狀煙草基因組上均有相近比例的覆蓋率,致使無法明確其染色體來源。該研究推測“Chromosome Nt17”的這種現象可能是由基因滲入或系譜特異染色體重排所導致。綜合上述分析,一方面說明在煙草基因組內可能存在染色體尺度的結構變異,這對揭示異源多倍體的物種起源與進化,基因組變異,多倍化現象等均有重要的研究意義:另一方面也表明,本研究的基因組測序和SNP鑒定可靠性良好,能夠準確地反映煙草基因組的序列結構特點,可用于進一步數據分析和發掘。
無論是在通過遺傳作圖群體進行連鎖分析(QTL定位)還是通過自然群體進行關聯分析(GWAS),SNP位點的數量都是關系到分析精度的關鍵指標,尤其是在GWAS研究中,SNP位點的數量還是決定能否進行有效候選基因預測的關鍵因素。本研究采用了RAD簡化基因組測序技術,該技術能夠有效壓縮基因組測序的數據量,從而大幅降低研究成本,將測序技術普及到更多的分子遺傳學研究中。雖然RAD技術鑒定到的SNP位點數量大幅低于全基因組測序,但仍然是一種高通量的基因組遺傳位點分型技術,且遠高于SSR等分子標記技術所能檢測到的位點數量。在QTL定位研究中,當上圖標記數量達到幾十kb,甚至僅幾kb時,群體大小就成為了影響定位精度的決定因素。如煙草NtEGY1和NtEGY2的基因圖位克隆研究中,所用遺傳連鎖圖譜的位點總數僅9.7 kb。因此本研究鑒定到的291 kb SNP位點已經足以支撐各類煙草性狀的精細定位:在GWAS研究中,需要在目標基因所處LD區段內檢測到一定數量的SNP位點。目前煙草基因組還缺少較為精準LD衰減距離,FRICANO A等利用7個SSR標記遺傳連鎖群,估算了普通煙草的平均LD距離約在1 cM以內,本研究將其折算成物理距離則約為1-3 Mb。根據本研究SNP位點的平均距離計算,在1 Mb的范圍內,檢測到的SNP數量達到了100個,因此即使基因組個別位置的LD衰減速度遠超平均值,本研究的SNP標記密度仍可有效錨定候選基因。且在GWAS研究中,供試材料的數量一般會在200個以上,因此在測序深度不變的情況下鑒定到的SNP位點必然會大幅增加,故在利用RAD技術進行煙草GWAS分析時,平均測序深度還可比本研究的1.01 x再降低,從而進一步減少測序成本。
目前SNP的檢測(Genotyping)方法日益豐富,如適用于高通量位點和群體的SNP芯片法(http://www.illumina.com/),中高通量基于質譜的iPLEX
GoldTM
Assay、
Mass
ARRAYTM(http://www.sequenom.com/iplex),目標序列捕捉或靶向測序(http://sequencing.roche.com),適合有限位點但群體規模較大的KASP標記法,適合少量位點和材料且對儀器設備要求較低的CASP/dCAPS標記、AS-PCR標記和SSCP檢測方法等。在完成SNP位點的開發后,相關研究可根據群體規模及位點通量需求選擇相應的檢測方法。
4結論
本研究對10份烤煙材料利用RAD技術進行了重測序,共鑒定到291770個SNP位點。煙草中SNP的變異類型以轉換為主,轉換與顛換比值為1.74。通過分析不同染色體上SNP的分布特點,發現在煙草基因組中17號染色體上半臂的SNP位點密度最高。基于高通量測序的RAD技術能夠為煙草遺傳研究提供足夠數量的SNP位點,還能大幅節省測序費用,在遺傳定位和基因發掘研究中具有良好的應用前景。
猜你喜歡
奧秘(創新大賽)(2023年3期)2023-05-06 01:48:20
中國煙草學報(2019年5期)2019-11-14 07:54:12
首都公共衛生(2019年5期)2019-05-21 01:08:34
浙江中西醫結合雜志(2017年2期)2017-01-12 18:23:59
新聞傳播(2016年3期)2016-07-12 12:55:34
當代化工研究(2016年9期)2016-03-20 16:22:08
自動化博覽(2014年6期)2014-02-28 22:32:15
聲屏世界(2014年6期)2014-02-28 15:18:09
西南學林(2013年2期)2013-11-12 12:58:54
中國煙草學報(2012年5期)2012-04-12 06:21:18
