棘腹蛙線粒體局部重復序列非排序聚類
2018-05-28 04:27:48曹躍夏云鄭渝池
四川動物 2018年3期
關(guān)鍵詞:差異
曹躍, 夏云, 鄭渝池*
(1. 中國科學院成都生物研究所兩棲爬行動物研究室,成都610041; 2. 中國科學院大學,北京100049)
動物線粒體基因組發(fā)生局部串聯(lián)重復后,涉及區(qū)域的序列具有多拷貝和大量插入缺失的特點,故難以排序。分析這類序列的演化時,可將序列進行聚類并以樹形結(jié)構(gòu)直觀地歸納和呈現(xiàn)序列間的相似性及差異。理論上講,可通過非排序比對方法(alignment-free comparison methods,AFM)計算序列間差異矩陣,由此得到聚類樹。但迄今未見此類評估和運用。線粒體基因組各位點間通常完全連鎖。如果基于AFM的重復區(qū)域序列聚類樹和非重復區(qū)域基因樹的拓撲結(jié)構(gòu)相近,才能被實際運用。基于什么樣的AFM和參數(shù)空間才能得到這樣的聚類樹則有待研究。
AFM始于20世紀80年代中期,其后不斷發(fā)展,已被應(yīng)用于不同領(lǐng)域,例如序列檢索(Hideetal. ,1994)、全基因組序列聚類(Simsetal. ,2009a;Renetal. ,2016)、宏基因組對比(Jiangetal. ,2012;Wangetal. ,2014)、調(diào)控序列演化和識別(Songetal. ,2013)、重組特別是重排序列分析(Vinga,2014;Zielezinskietal. ,2017)。AFM主要分為2大類:一類不對序列進行拆分,如計算全長序列間共享信息(Vinga & Almeida,2003;Vinga,2013);另一類將序列分解為特定長度(k)的子序列集作為對比時的數(shù)據(jù)來源(Ulitskyetal. ,2006;Almeida,2013)。后者在準確性、計算資源占用、序列差異程度等方面有較好表現(xiàn)(Haubold,2013;Bernardetal. ,2016;Zielezinskietal. ,2017)。
序列間差異越小,其k長度子序列集的相似程度越高,可通過不同特征進行度量。一類方法基于子序列集中獨特序列的計數(shù),如Co-phylog算法(Yi & Jin,2013),有對該計算進行校正的版本,如CVTree算法(Qietal. ,2004)。另一類常見方法考查某條序列是否在子序列集中出現(xiàn),如Yule算法(Luetal. ,2017)。k值直接決定每條序列的子序列集,一組序列在不同k值下生成不同的距離矩陣,為一重要參數(shù),其在不同應(yīng)用中的適宜值域不同。在進行氨基酸序列對比時,有工作提示k值為2~6時可以得到穩(wěn)定的結(jié)果(H?hletal. ,2006;H?hl & Ragan,2007)。進行核酸對比時,k值可設(shè)為8~10(Chanetal. ,2014);9~14可用于聚類樹構(gòu)建(Simsetal. ,2009b;Junetal. ,2010;Wangetal. ,2014)。一般來講,當序列差異較大時,k值應(yīng)較小;反之,當序列相似性高時,k值應(yīng)較大(Wuetal. ,2005;Simsetal. ,2009b;Zielezinskietal. ,2017)。
脊椎動物線粒體基因tRNA簇trnW、trnA、trnN、OL、trnC、trnY是發(fā)生串聯(lián)重復的一個熱點區(qū)域,習慣上稱“WANCY”區(qū),其中,OL為輕鏈復制起點。重復后會發(fā)生基因的隨機丟失,故該區(qū)域往往含有同一基因的功能序列和殘基(San Mauroetal. ,2006;Fonseca & Harris,2008)。棘腹蛙Quasipaaboulengeri線粒體基因組存在這樣的重復和隨機丟失,其WANCY序列依功能基因的排列可分為Ⅰ~Ⅳ 4種類型(Xiaetal. ,2016)。Ⅰ型序列和其他類型明顯不同。重排區(qū)外蛋白編碼序列的線粒體基因樹也顯示Ⅱ~Ⅳ型聚為一支,是一次串聯(lián)重復后的不同隨機丟失形式,長度600~700 bp。其中,Ⅲ型和Ⅳ型的關(guān)系更近,但Ⅲ型有2次起源,分為Ⅲa和Ⅲb,后者和Ⅳ型關(guān)系更近(Xiaetal. ,2016)。這也表明功能基因的排列順序不足以反映這些重排序列的相似性和差異程度。
選取19號有代表性的Ⅱ~Ⅳ型棘腹蛙個體,采用基于k長度子序列集的AFM對其WANCY序列進行聚類,以其1 518 bp蛋白編碼序列線粒體基因樹為參照,計算兩者間Robinson-Foulds拓撲結(jié)構(gòu)距離(Robinson & Foulds,1981),并考查AFM聚類樹能否反映Ⅲa型、Ⅲb型和Ⅳ型間的關(guān)系,探尋在該條件下有更好表現(xiàn)的AFM和k值范圍,以期為將AFM引入動物線粒體串聯(lián)重復序列的演化研究提供幫助。
1 材料和方法
1.1 樣品和DNA序列
樣品選自作者所在研究團隊前期發(fā)表的一項棘腹蛙WANCY序列演化研究的樣本(Xiaetal. ,2016)。選擇依據(jù)為具有代表性和穩(wěn)定的基因樹拓撲結(jié)構(gòu)。19號樣品來自Ⅱ~Ⅳ型,Ⅱ型作為外群。DNA序列來自2個片段:1個片段包括WANCY及其兩翼的nad2(115 bp)和cox1(565 bp)基因部分序列;另1個片段為長度838 bp的cytb基因部分序列。前者全部和后者中12條序列下載自GenBank,登錄號:KX571520、KX571524、KX571530、KX571533、KX571553、KX571557~ KX571560、KX571565、KX571568、KX571570、KX571571、KX571574、KX571583、KX571586、KX571588、KX571591、KX571592、KX571838、KX571843、KX571844、KX571850、KX571860、KX571861、KX571865、KX571876、KX571880、KX571886、KX571892、KX571898(Xiaetal. ,2016)。另外7條cytb基因序列為實驗獲得,所用PCR引物為CYTBF-3(5’-ACCTCCCCGCTCCAGCAAATCTA-3’)和CYTBR-3(5’-CCGATGGTGACGAATGGGTCTTC-3’),退火溫度52 ℃,GenBank登錄號:MG799837~MG799843。蛋白編碼序列的排序采用Clustal X 2.1(Larkinetal. ,2007)。
1.2 蛋白編碼序列基因樹和WANCY序列AFM聚類樹的構(gòu)建和比較
蛋白編碼序列基因樹的構(gòu)建采用常用的最大似然法(ML),使用RAxML 8.2.10(Stamatakis,2014)。3個基因片段nad2、cox1、cytb被合并為1個數(shù)據(jù)集。由于是種下水平的分析,未嘗試根據(jù)基因或密碼子位點對數(shù)據(jù)集進行分區(qū),以避免可能的過參數(shù)化。在RAxML已有DNA序列演化模型中,依據(jù)貝葉斯信息準則使用PartitionFinder 2.1.1(Lanfearetal. ,2016)選擇最適模型。RAxML分析包括1 000次獨立推斷得出最優(yōu)ML樹,自展法1 000次重復計算其支持率。
WANCY序列AFM聚類使用CAFE 071017(Luetal. ,2017)。該軟件是對基于k長度子序列集AFM的最新整合。作為將AFM引入線粒體串聯(lián)重復序列聚類的探索,分析采用其編譯的全部28種可使用普通個人計算機運算的AFM。這些算法的名稱與CAFE一致。采用默認設(shè)置逐一計算得到k值為4、6、8、10、12、14、16、18、20時的距離矩陣。隨后使用Phylip 3.695(Felsenstein,1989)中的鄰接法將這些距離矩陣轉(zhuǎn)換成聚類樹。
基因樹和AFM聚類樹間Robinson-Foulds距離的計算使用DendroPy 4.3.0(Sukumaran & Holder,2010)。Python庫使用Newick格式樹文件,格式轉(zhuǎn)換在FigTree 1.42(http://tree.bio.ed.ac.uk/software/figtree/)中進行。另外,記錄AFM聚類樹是否將序列劃分為Ⅱ、Ⅲa、Ⅲb、Ⅳ支系,以及是否呈現(xiàn)和基因樹相同的(Ⅲa,(Ⅲb,Ⅳ))支系關(guān)系。
2 結(jié)果
19條蛋白編碼序列排序未發(fā)現(xiàn)插入或缺失。所得數(shù)據(jù)集含1 518個位點,其中可變位點116個,簡約信息位點81個。RAxML中適合該數(shù)據(jù)集的模型為GTR+I+G。所得ML樹拓撲結(jié)構(gòu)總體穩(wěn)定,平均自展支持率為89(圖1)。
19條WANCY序列長度在583~695 bp之間。28種AFM與k值共產(chǎn)生252種組合。有7種AFM可以完成全部組合的計算。普通個人計算機無法滿足部分組合(k值為12、14、16、18、20)的計算量,反映了k值與計算資源需求間的一定正相關(guān)。另外,有3個組合得到明顯無意義的聚類樹,所有序列無支長或呈梳狀,分別為Co-phylog算法(k值為4)、Hamming算法(k值為18)、Russel算法(k值為8),無法總結(jié)出規(guī)律。最終得到141個組合的聚類樹用于后續(xù)分析,它們與ML樹的Robinson-Foulds距離及主要拓撲關(guān)系比較總結(jié)見表1。
141個AFM聚類樹與ML樹的Robinson-Foulds距離介于4~24,這表明沒有得到和ML樹拓撲結(jié)構(gòu)完全相同的AFM聚類樹,兩者最相近的情況為2個節(jié)點(11.8%)的差異,即Robinson-Foulds距離等于4。得到距離為4的組合共16個(11.3%),其中14個來自幾乎所有的基于特定序列是否在子序列集中出現(xiàn)的算法,另外2個來自基于子序列集中獨特序列計數(shù)的Canberra算法。這16個組合來自14種算法,其中12種/個算法/組合的k值為8,Chisq算法的2個組合k值為4、6,Canberra算法的2個組合k值為4、20。得到距離6和8的組合各自為66個和20個,分別占全部組合的46.8%和14.2%。這些和ML樹更接近的聚類占全部結(jié)果的72.3%,但并不是所有的AFM都可得出。D2star、CVTree、Ch、FFP算法所得聚類樹和ML樹的距離為10~24,平均為18,占全部結(jié)果的17.0%。
可以將19條WANCY序列聚為Ⅱ、Ⅲa、Ⅲb、Ⅳ4支的組合共121個,占85.8%。大多數(shù)算法在不同k值下均可得到這樣的結(jié)果,例如Ma和Canberra算法。但來自D2star、CVTree、Ch算法的組合無法得出這樣的聚類。呈現(xiàn)和ML樹相同(Ⅲa,(Ⅲb,Ⅳ))支系關(guān)系的15個組合占全部組合的10.6%。這15個組合來自13種算法,其中11種算法/組合的k值為4,另外2種算法Hamming和Canberra的各2個組合k值分別為4、20。
得到和ML樹最小距離的16個組合條件與發(fā)現(xiàn)ML樹主要支系關(guān)系的15個組合條件的重疊度不高(表1)。主要表現(xiàn)為,很多方法中k值為8時距離最小,k值為4時呈現(xiàn)關(guān)系(Ⅲa,(Ⅲb,Ⅳ))。兩大類條件間僅重疊2個組合,即Canberra算法設(shè)k值為4和20時,可認為是本工作中的最理想AFM分析條件。2個條件下所得聚類樹拓撲結(jié)構(gòu)相同,前者作為示例見圖1,其和ML樹的拓撲關(guān)系的區(qū)別僅存在于支系Ⅳ內(nèi),可能與ML樹該支內(nèi)部分節(jié)點的支持率不高有一定關(guān)聯(lián)。
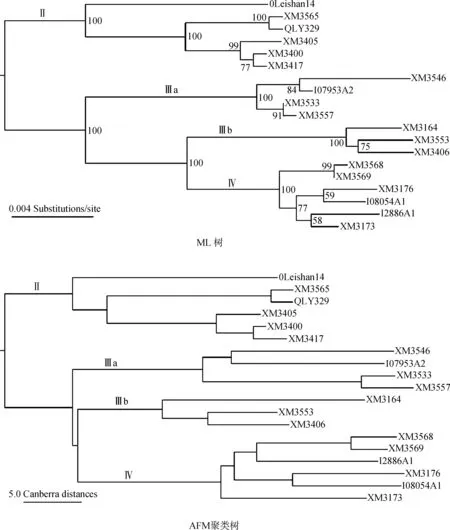
圖1 蛋白編碼序列最大似然樹和運用Canberra算法設(shè)k值為4所得WANCY序列非排序比對樹Fig. 1 The Maximum Likelyhood tree of the protein coding sequences and the Canberra alignment-free comparison method tree of the WANCY sequences using a k value of 4
ML樹節(jié)點旁數(shù)值為自展支持率; 蛋白編碼序列和WANCY序列均使用該號個體的編號
Numbers beside nodes of the ML tree are bootstrap supporting values; tip labels are lab numbers of the 19 individuals
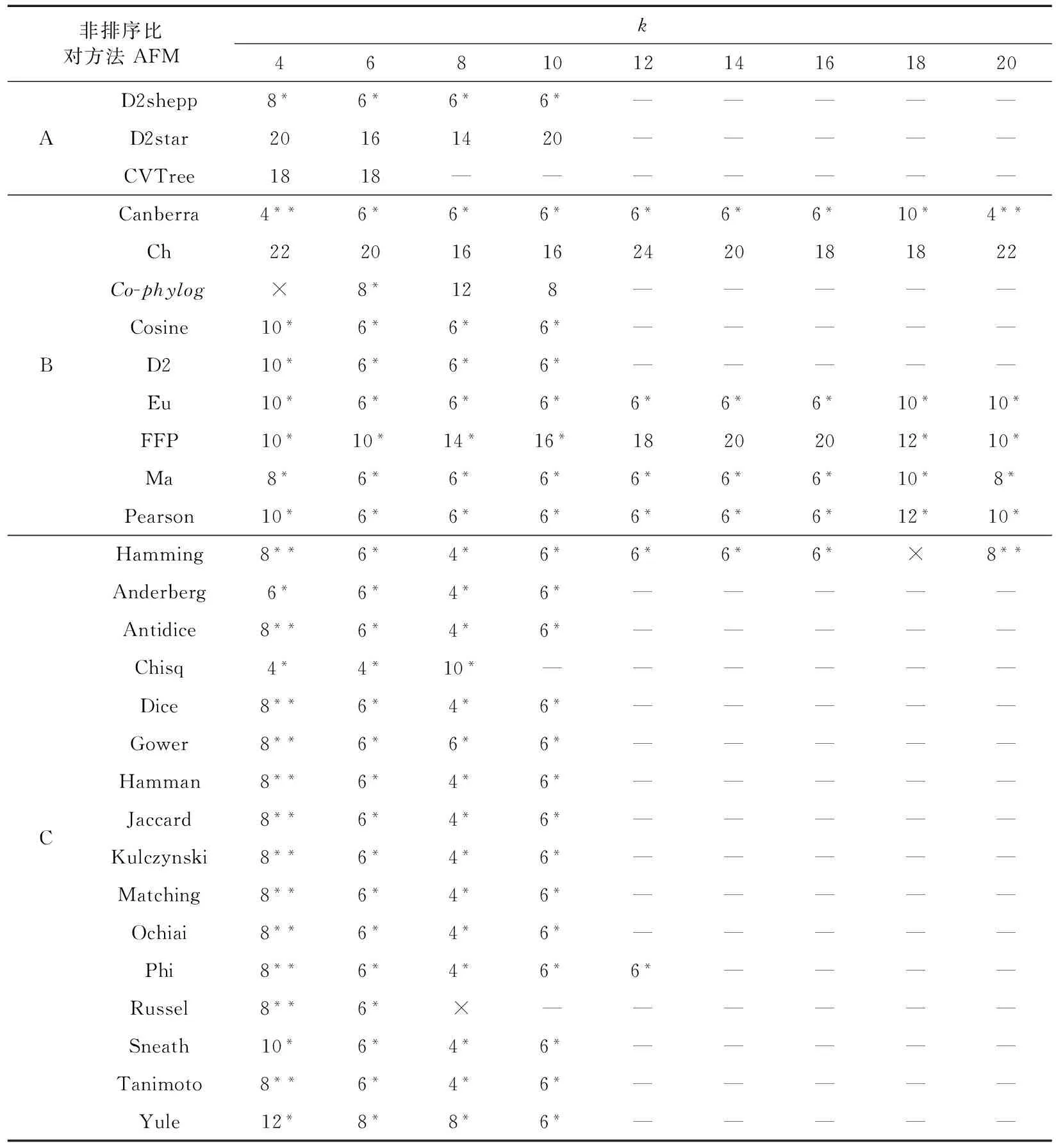
表1 最大似然樹和不同k值下非排序比對聚類樹Robinson-Foulds距離及主要拓撲關(guān)系比較Table 1 Robinson-Foulds distances and topology comparison between the Maximum Likelyhood treeand alignment-free comparison trees obtained using various k values
注: A. 基于校正后子序列集中獨特序列的計數(shù); B. 基于子序列集中獨特序列的計數(shù); C. 基于序列是否在子序列集中出現(xiàn)的計數(shù);*AFM聚類樹將序列劃分為Ⅱ、Ⅲa、Ⅲb、Ⅳ 4個支系;**AFM聚類樹呈現(xiàn)和ML樹相同的(Ⅲa,(Ⅲb,Ⅳ))支系關(guān)系; × 明顯錯誤的結(jié)果; — 運算無法在i7-6700HQ 2.60 GHz CPU、8 GB RAM配置的個人電腦上完成
Notes: A. methods using adjustedk-mer counts; B. methods based onk-mer counts only; C, methods based on presence/absence ofk-mers;*AFM tree clustering sequences into clades Ⅱ, Ⅲa, Ⅲb, and Ⅳ;**AFM tree shows a (Ⅲa, (Ⅲb, Ⅳ)) relationship observed also on the ML tree; × apparently strange result; — computation can’t be finished in a reasonable time on a laptop with i7-6700HQ 2.60 GHz CPU and 8 GB RAM
3 討論
本工作表明,AFM聚類樹可以被用于顯示動物線粒體串聯(lián)重復序列的關(guān)系和相似性,并達到較為理想的效果。對于棘腹蛙一組種下變異水平的WANCY序列,大多數(shù)(85.8%)算法和k值組合都可以成功地將其聚為Ⅱ、Ⅲa、Ⅲb、Ⅳ四大支。可見這些基于k長度子序列集的算法總體上對該系統(tǒng)中的主要序列差異有很好的判別能力。而且,很多方法都可以獲得和蛋白編碼序列樹相近的結(jié)果。所測試的28種算法中,半數(shù)可在特定k值下產(chǎn)生和ML樹拓撲結(jié)構(gòu)非常接近、相差僅2個節(jié)點的聚類樹,也能呈現(xiàn)前述序列差異較大的支系間的關(guān)系。最理想條件下,Canberra算法聚類樹和ML樹的差別僅存在于支系Ⅳ內(nèi)的2個節(jié)點,有效地歸納和呈現(xiàn)了序列間的相似性及差異。這樣的表現(xiàn)也提示了此類算法具有被運用于其他動物線粒體串聯(lián)重復序列系統(tǒng)的潛力。
但是,不同AFM在本工作中的表現(xiàn)可以有很大的差異。和其他算法相比,D2star、CVTree、Ch、FFP算法所得聚類樹無論是在大支關(guān)系還是在細節(jié)上,都和ML樹有較大的差別。例如,不同k值所得Ch樹和ML樹的平均Robinson-Foulds距離為20,表明兩者間的大多數(shù)節(jié)點都不同。這提示可能不是所有的AFM都適用于動物線粒體基因組復制重排區(qū)域序列的聚類。上述4種算法都基于子序列集中獨特序列的計數(shù),但表現(xiàn)最好的Canberra算法同屬此類,不支持根據(jù)類型選擇算法。這些結(jié)果提示了在不同復制重排系統(tǒng)中進行類似評估的必要性。
針對不同評估標準,不同算法內(nèi)存在可以得出最理想聚類的特定k值,而且算法間表現(xiàn)出較高的一致性,具有規(guī)律。絕大多數(shù)得出最小Robinson-Foulds距離的組合中,k值為8。而在幾乎全部得出和ML樹支系關(guān)系一致的組合中,k值為4。Robinson-Foulds距離反映拓撲結(jié)構(gòu)間的全部節(jié)點差異,不考慮節(jié)點位置所代表的序列間差異程度,顯示在棘腹蛙WANCY系統(tǒng)中k值為8時可以很好地顯示序列間的關(guān)系。作為經(jīng)驗數(shù)據(jù),這與基于模擬數(shù)據(jù)所得出的適合于DNA序列聚類的8~10的k值域一致(Chanetal. ,2014)。大支間拓撲結(jié)構(gòu)代表差異相對較大的序列間關(guān)系,本文所得適合解析這樣關(guān)系的k值為4。該現(xiàn)象的實質(zhì)是,不同的k值適合解析不同差異程度的序列間關(guān)系,存在權(quán)衡。這樣的權(quán)衡已經(jīng)在AFM中被發(fā)現(xiàn)。總的來講,較小的k值適合聚類差異程度高的序列,較大的k值適合更相似序列(Bonham-Carteretal. ,2013),需要根據(jù)研究目的進行設(shè)置。而本研究結(jié)果表明,即使是種下水平的研究,也可能需要考慮這種權(quán)衡。如果聚類的目的是作為輔助手段展示序列間的異同,可以考慮對數(shù)據(jù)進行分層次的分析。例如,采用較小的k值聚類得到大支系間關(guān)系,在各支系內(nèi)的聚類則設(shè)定較大的k值,再整合展示結(jié)果。另外值得一提的是,Canberra和Hamming算法在k值分別為4和20時得到較理想結(jié)果,但原因有待解析。
總結(jié)以上,本工作例證AFM可有效聚類蛙類WANCY序列,所得算法k值組合對類似系統(tǒng)有借鑒意義,揭示了AFM對于動物線粒體復制重排序列演化研究的價值。對于同一組數(shù)據(jù),不同算法的表現(xiàn)可能迥異。WANCY區(qū)的主要特點是tRNA基因復制后隨機假基因化并導致重排,對其他類型的復制重排區(qū)也可開展評估以辨識算法。k值的設(shè)定與序列差異程度相關(guān),根據(jù)聚類目的可嘗試對數(shù)據(jù)進行分層分析。
致謝:感謝中國科學院成都生物研究所戴強老師在數(shù)據(jù)分析中給予的幫助。
:
Almeida JS. 2013. Sequence analysis by iterated maps, a review[J]. Briefings in Bioinformatics, 15(3): 369-375.
Bernard G, Chan CX, Ragan MA. 2016. Alignment-free microbial phylogenomics under scenarios of sequence divergence, genome rearrangement and lateral genetic transfer[J]. Scientific Reports, 6: 28970.
Bonham-Carter O, Steele J, Bastola D. 2013. Alignment-free genetic sequence comparisons: a review of recent approaches by word analysis[J]. Briefings in Bioinformatics, 15(6): 890-905.
Chan CX, Bernard G, Poirion O,etal. 2014. Inferring phylogenies of evolving sequences without multiple sequence alignment[J]. Scientific Reports, 4: 6504.
Felsenstein J. 1989. PHYLIP-phylogeny inference package (version 3.2)[J]. Cladistics, 5(2): 164-166.
Fonseca MM, Harris DJ. 2008. Relationship between mitochondrial gene rearrangements and stability of the origin of light strand replication[J]. Genetics and Molecular Biology, 31(2): 566-574.
Haubold B. 2013.Alignment-free phylogenetics and population genetics[J]. Briefings in Bioinformatics, 15(3): 407-418.
Hide W, Burke J, Da Vison DB. 1994. Biological evaluation of d2, an algorithm for high-performance sequence comparison[J]. Journal of Computational Biology, 1(3): 199-215.
H?hl M, Ragan MA. 2007. Is multiple-sequence alignment required for accurate inference of phylogeny?[J]. Systematic Biology, 56(2): 206-221.
H?hl M, Rigoutsos I, Ragan MA. 2006. Pattern-based phylogenetic distance rstimation and tree reconstruction[J]. Evolutionary Bioinformatics Online, 2(1): 359-375.
Jiang B, Song K, Ren J,etal. 2012. Comparison of metagenomic samples using sequence signatures[J]. BMC Genomics, 13: 730.
Jun SR, Sims GE, Wu GA,etal. 2010. Whole-proteome phylogeny of prokaryotes by feature frequency profiles: an alignment-free method with optimal feature resolution[J]. Proceedings of the National Academy of Sciences, 107(1): 133-138.
Lanfear R, Frandsen PB, Wright AM,etal. 2016. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses[J]. Molecular Biology and Evolution, 34(3): 772-773.
Larkin MA, Blackshields G, Brown NP,etal. 2007. Clustal W and Clustal X version 2.0[J]. Bioinformatics, 23(21): 2947-2948.
Lu YY, Tang K, Ren J,etal. 2017. CAFE: aCcelerated Alignment-FrEe sequence analysis[J]. Nucleic Acids Research, 45: W554-W559.
Qi J, Luo H, Hao B. 2004. CVTree: a phylogenetic tree reconstruction tool based on whole genomes[J]. Nucleic Acids Research, 32: W45-W47.
Ren J, Song K, Deng M,etal. 2016. Inference of Markovian properties of molecular sequences from NGS data and applications to comparative genomics[J]. Bioinformatics, 32(7): 993-1000.
Robinson DF, Foulds LR. 1981. Comparison of phylogenetic trees[J]. Mathematical Biosciences, 53(1-2): 131-147.
San Mauro D, Gower DJ, Zardoya R,etal. 2006. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome [J]. Molecular Biology and Evolution, 23(1): 227-234.
Sims GE, Jun SR, Wu GA,etal. 2009a. Whole-genome phylogeny of mammals: evolutionary information in genic and nongenic regions[J]. Proceedings of the National Academy of Sciences, 106(40): 17077-17082.
Sims GE, Jun SR, Wu GA,etal. 2009b. Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions[J]. Proceedings of the National Academy of Sciences, 106(8): 2677-2682.
Song K, Ren J, Reinert G,etal. 2013. New developments of alignment-free sequence comparison: measures, statistics and next-generation sequencing[J]. Briefings in Bioinformatics, 15(3): 343-353.
Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies[J]. Bioinformatics, 30(9): 1312-1313.
Sukumaran J, Holder MT. 2010. DendroPy: a Python library for phylogenetic computing[J]. Bioinformatics, 26(12): 1569-1571.
Ulitsky I, Burstein D, Tuller T,etal. 2006. The average common substring approach to phylogenomic reconstruction [J]. Journal of Computational Biology, 13(2): 336-350.
Vinga S, Almeida J. 2003. Alignment-free sequence comparison-a review[J]. Bioinformatics, 19(4): 513-523.
Vinga S. 2013.Information theory applications for biological sequence analysis[J]. Briefings in Bioinformatics, 15(3): 376-389.
Vinga S. 2014. Alignment-free methods in computational biology[J]. Briefings in Bioinformatics, 15(3): 341-342.
Wang Y, Liu L, Chen L,etal. 2014. Comparison of metatranscriptomic samples based onk-tuple frequencies[J]. PLoS ONE, 9(1): e84348. DOI: 10.1371/journal.pone.0084348.
Wu TJ, Huang YH, Li LA. 2005. Optimal word sizes for dissimilarity measures and estimation of the degree of dissimilarity between DNA sequences[J]. Bioinformatics, 21(22): 4125-4132.
Xia Y, Zheng Y, Murphy RW,etal. 2016. Intraspecific rearrangement of mitochondrial genome suggests the prevalence of the tandem duplication-random loss (TDLR) mechanism inQuasipaaboulengeri[J]. BMC Genomics, 17: 965.
Yi H, Jin L. 2013.Co-phylog: an assembly-free phylogenomic approach for closely related organisms[J]. Nucleic Acids Research, 41(7): e75. DOI: 10.1093/nar/gkt003.
Zielezinski A, Vinga S, Almeida J,etal. 2017. Alignment-free sequence comparison: benefits, applications, and tools[J]. Genome Biology, 18(1): 186.
猜你喜歡
英語世界(2023年10期)2023-11-17 09:19:16
汽車實用技術(shù)(2022年10期)2022-06-09 11:16:58
音樂探索(2022年2期)2022-05-30 21:01:37
收藏界(2019年3期)2019-10-10 03:16:40
小天使·一年級語數(shù)英綜合(2019年8期)2019-08-27 02:23:00
中國特種設(shè)備安全(2018年11期)2019-01-08 02:08:32
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
中國非營利評論(2017年1期)2017-11-09 03:09:10
海外華文教育(2017年8期)2017-11-07 04:42:02
現(xiàn)代語文(2016年21期)2016-05-25 13:13:50
