HPLC法同時測定余甘子中5種成分的含量及主成分、聚類分析Δ
2018-06-12 08:18:12李琦裴河歡李靜羅亞虹桂林市食品藥品檢驗所廣西桂林541012欽州市中醫醫院科研科廣西欽州535000欽州市中醫藥研究所廣西欽州535000
中國藥房 2018年11期
關鍵詞:質量
李琦,裴河歡,李靜,羅亞虹(1.桂林市食品藥品檢驗所,廣西桂林541012;2.欽州市中醫醫院科研科,廣西欽州 535000;3.欽州市中醫藥研究所,廣西欽州 535000)
余甘子為大戟科植物余甘子(Phyllanthus emblicaL.)的干燥成熟果實,異名油甘子、牛甘子、喉甘子、魚木果等,是一味傳統民族藥,主要分布于福建、廣東、廣西等地。其以果實入藥,主治血熱血瘀、消化不良、腹脹、咳嗽、喉痛等癥[1-2]。現代藥理學研究表明,余甘子中富含的多元酚類化合物具有抗氧化作用,能減輕自由基對機體的損傷,緩解疲勞[3-4]。文獻[5-7]報道了同時測定余甘子中3~4種多元酚類化合物的含量。為提高余甘子質量控制標準,筆者采用高效液相色譜(HPLC)法同時測定廣西余甘子中5種多元酚類化合物的含量,并對廣西12個采集地余甘子中所含5種多元酚類化合物的含量進行主成分分析和聚類分析[8-14],將復雜的多指標轉化為少數代表性的綜合指標,從而識別藥材質量的優劣和產地的規律性,為余甘子藥材質量控制和綜合評價提供科學依據。
1 材料
1.1 儀器
Agilent1260 InfinityⅡHPLC儀,包括G7115A二極管陣列寬波長范圍檢測器、G7129A自動進樣器、G7111B四元梯度泵(美國Agilent公司);XS205DU電子分析天平(瑞士Mettler-Toledo公司);101-1-BS-Ⅱ鼓風干燥箱(上海躍進醫療器械有限公司);DTA-42超聲波清洗機[鼎泰(湖北)生化科技設備制造有限公司];Simplicity超純水系統(德國Merck Millipore公司)。
1.2 試劑
沒食子酸對照品(批號:110831-201605,純度:90.8%)、柯里拉京對照品(批號:111623-200301,純度:100%)、鞣花酸對照品(批號:111959-201602,純度:89.3%)均購自中國食品藥品檢定研究院;沒食子兒茶素對照品(批號:PRF7081801,純度:98%)、訶子聯苯酸對照品(批號:PRF8032222,純度:92%)均購自成都普瑞法科技開發有限公司;乙腈、磷酸為色譜純,其余試劑均為分析純,水為超純水。
1.3 藥材
藥材來源見表1。所有樣品經挑選,為飽滿、質實、無蟲霉者。經廣西中醫藥大學藥學院劉壽養副教授鑒定均為大戟科植物余甘子(Phyllanthus emblicaL.)的新鮮成熟果實。將挑選后的樣品用超純水洗凈,干燥,去核,粉碎制成直徑為0.5~0.8 cm的粗顆粒,充分混合均勻,再按四分法取樣,每份約200 g,備用。
生品:取余甘子粗顆粒,粉碎,過四號篩,混合均勻,備用。

表1 藥材來源Tab 1 Sources of P.emblica
2 方法與結果
2.1 色譜條件
色譜柱:Agilent Eclipse XDB(250 mm×4.6 mm,5 μm);流動相:乙腈(A)-0.1%磷酸水溶液(B),梯度洗脫(0~10 min,4%A;10~11 min,4%→6%A;11~25 min,6%→13%A;25~35 min,13%→14%A;35~36 min,14%→15%A;36~50 min,15%→17%A;50~60 min,17%→4%A);流速:1.0 mL/min;檢測波長:220 nm;柱溫:25 ℃;進樣量:20 μL。
2.2 溶液的制備
2.2.1 對照品貯備液 ①精密稱取沒食子酸對照品10.98 mg,置于50 mL棕色量瓶中,加50%甲醇溶解并稀釋至刻度,搖勻,得質量濃度為199.4 μg/mL的對照品貯備液A。精密吸取對照品貯備液A 5 mL,置于25 mL棕色量瓶中,加50%甲醇溶解并稀釋至刻度,搖勻,得質量濃度為39.88 μg/mL的對照品貯備液B。②精密稱取沒食子兒茶素對照品10.19 mg,置于100 mL棕色量瓶中,加甲醇溶解并稀釋至刻度,搖勻,精密吸取5 mL,置于25 mL棕色量瓶中,加甲醇溶解并稀釋至刻度,搖勻,得質量濃度為19.97 μg/mL的對照品貯備液C。精密吸取對照品貯備液C 5 mL,置于50 mL棕色量瓶中,加甲醇溶解并稀釋至刻度,搖勻,得質量濃度為1.997 μg/mL的對照品貯備液D。③精密稱取柯里拉京對照品15.11mg,置于100 mL棕色量瓶中,加甲醇溶解并稀釋至刻度,搖勻,得質量濃度為151.1 μg/mL的對照品貯備液E。④精密稱取訶子聯苯酸對照品20.04 mg,置于50 mL棕色量瓶中,加甲醇溶解并稀釋至刻度,搖勻,得質量濃度為368.7 μg/mL的對照品貯備液F。⑤精密稱取鞣花酸對照品16.55 mg,置于50 mL棕色量瓶中,加甲醇溶解并稀釋至刻度,搖勻,得質量濃度為295.6 μg/mL的對照品貯備液G。精密吸取對照品貯備液G 5 mL,置于50 mL棕色量瓶中,加甲醇溶解并稀釋至刻度,搖勻,得質量濃度為29.56 μg/mL的對照品貯備液H。
2.2.2 混合對照品溶液 分別精密吸取“2.2.1”項下對照品貯備液B、D、E、F、H各1 mL,置于同一10 mL棕色量瓶中,甲醇定容,制成含沒食子酸、沒食子兒茶素、柯里拉京、訶子聯苯酸、鞣花酸質量濃度分別為3.988、0.199 7、15.11、36.87、2.956 μg/mL的混合對照品溶液。
2.2.3 供試品溶液 取“1.3”項下生品約0.1 g,精密稱定,置于具塞錐形瓶中,精密加入50%甲醇50 mL,稱定質量,超聲(功率:900 W,頻率:40 kHz)30 min,放冷,再稱定質量,用50%甲醇補足減失的質量,搖勻,經0.45μm微孔濾膜濾過,取續濾液,即得。
2.3 系統適用性試驗
取“2.2.2”項下混合對照品溶液和“2.2.3”項下供試品溶液(編號S5)適量,按“2.1”項下色譜條件進樣測定,記錄色譜圖。結果,在該色譜條件下,各待測成分間與雜質峰間均能達到基線分離,分離度>1.5,其他成分對待測成分的測定無干擾,理論板數以沒食子酸、沒食子兒茶素、柯里拉京、訶子聯苯酸、鞣花酸峰計均>3 000,色譜圖見圖1。
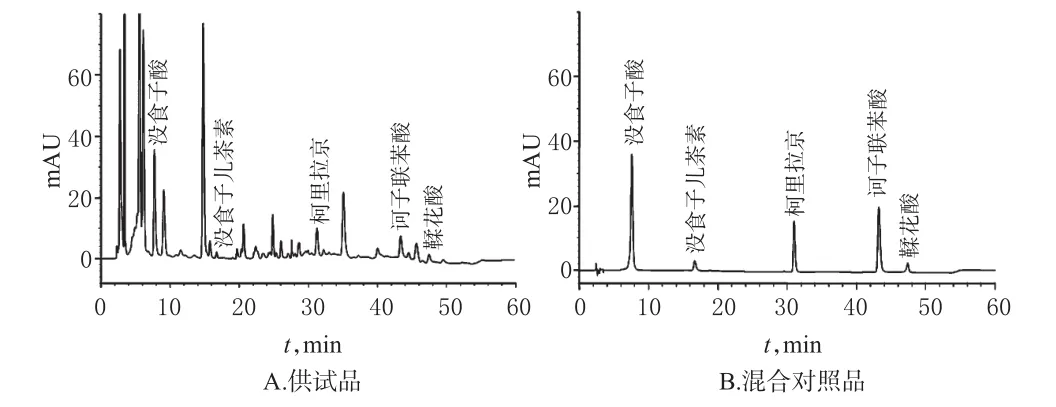
圖1 高效液相色譜圖Fig 1 HPLC chromatograms
2.4 線性關系考察
精密量取“2.2.1”項下對照品貯備液A 0.5、1.0、2.0、4.0、8.0 mL,對照品貯備液C 0.4、0.8、1.6、3.2、6.4 mL,對照品貯備液E 1.5、3.0、6.0、12.0、15.0 mL,對照品貯備液F 2.0、4.0、8.0、10.0、12.0 mL,對照品貯備液G 0.5、1.0、2.0、4.0、8.0 mL,分別置于50 mL棕色量瓶中,加甲醇定容,制成系列混合對照品溶液。精密量取上述系列混合對照品溶液各20 μL,按“2.1”項下色譜條件進樣測定,記錄峰面積。以沒食子酸、沒食子兒茶素、柯里拉京、訶子聯苯酸、鞣花酸質量濃度為橫坐標(x)、峰面積為縱坐標(y)進行線性回歸。線性關系考察結果見表2。
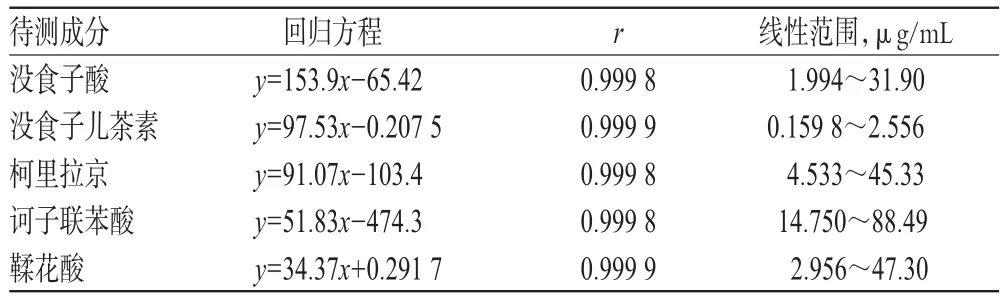
表2 線性關系考察結果Tab 2 Results of linear range investigation
2.5 檢測限與定量限考察
取“2.2.2”項下混合對照品溶液適量,倍比稀釋,按“2.1”項下色譜條件連續進樣測定6次,記錄峰面積。當信噪比為3∶1時,得檢測限;當信噪比為10∶1時,得定量限。結果,沒食子酸、沒食子兒茶素、柯里拉京、訶子聯苯酸和鞣花酸的檢測限分別為0.025 6、0.027 1、0.052 9、0.186 7、0.133 1 μg/mL,定量限分別為0.085 1、0.089 3、0.170 6、0.615 2、0.441 9 μg/mL。
2.6 精密度試驗
取“2.2.2”項下混合對照品溶液適量,按“2.1”項下色譜條件連續進樣測定6次,記錄峰面積,計算日內精密度;連續進樣測定3 d,計算日間精密度。結果,沒食子酸、沒食子兒茶素、柯里拉京、訶子聯苯酸、鞣花酸峰面積的日內精密度RSD分別為0.21%、0.88%、0.14%、0.20%、0.64%(n=6),日間精密度RSD分別為0.26%、1.42%、0.25%、0.24%、0.67%(n=6),表明儀器精密度良好。
2.7 穩定性試驗
取“2.2.3”項下供試品溶液(編號S5)適量,分別于室溫下放置0、2、4、8、10、12、24 h時按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,沒食子酸、沒食子兒茶素、柯里拉京、訶子聯苯酸、鞣花酸峰面積的RSD分別為0.45%、0.63%、0.27%、0.57%、0.60%(n=7),表明供試品溶液在室溫放置24 h內穩定性良好。
2.8 重復性試驗
精密稱取編號S5的生品約0.1 g,按“2.2.3”項下方法制備供試品溶液,共6份,再按“2.1”項下色譜條件進樣測定,記錄峰面積并扣除生品(編號S5)含水量后,再計算含量。結果,沒食子酸、沒食子兒茶素、柯里拉京、訶子聯苯酸、鞣花酸含量平均值分別為2.314 4、0.109 1、6.138 6、12.394 5、1.938 1 mg/g,RSD 分別為 0.76%、1.38%、0.55%、0.84%、1.02%(n=6),表明本方法重復性良好。
2.9 加樣回收率試驗
精密稱取已知含量的“1.3”項下生品(編號S5)約0.05 g,共6份,分別加入一定體積的“2.2.1”項下對照品貯備液,按“2.2.3”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并扣除生品(編號S5)含水量計算,結果見表3。
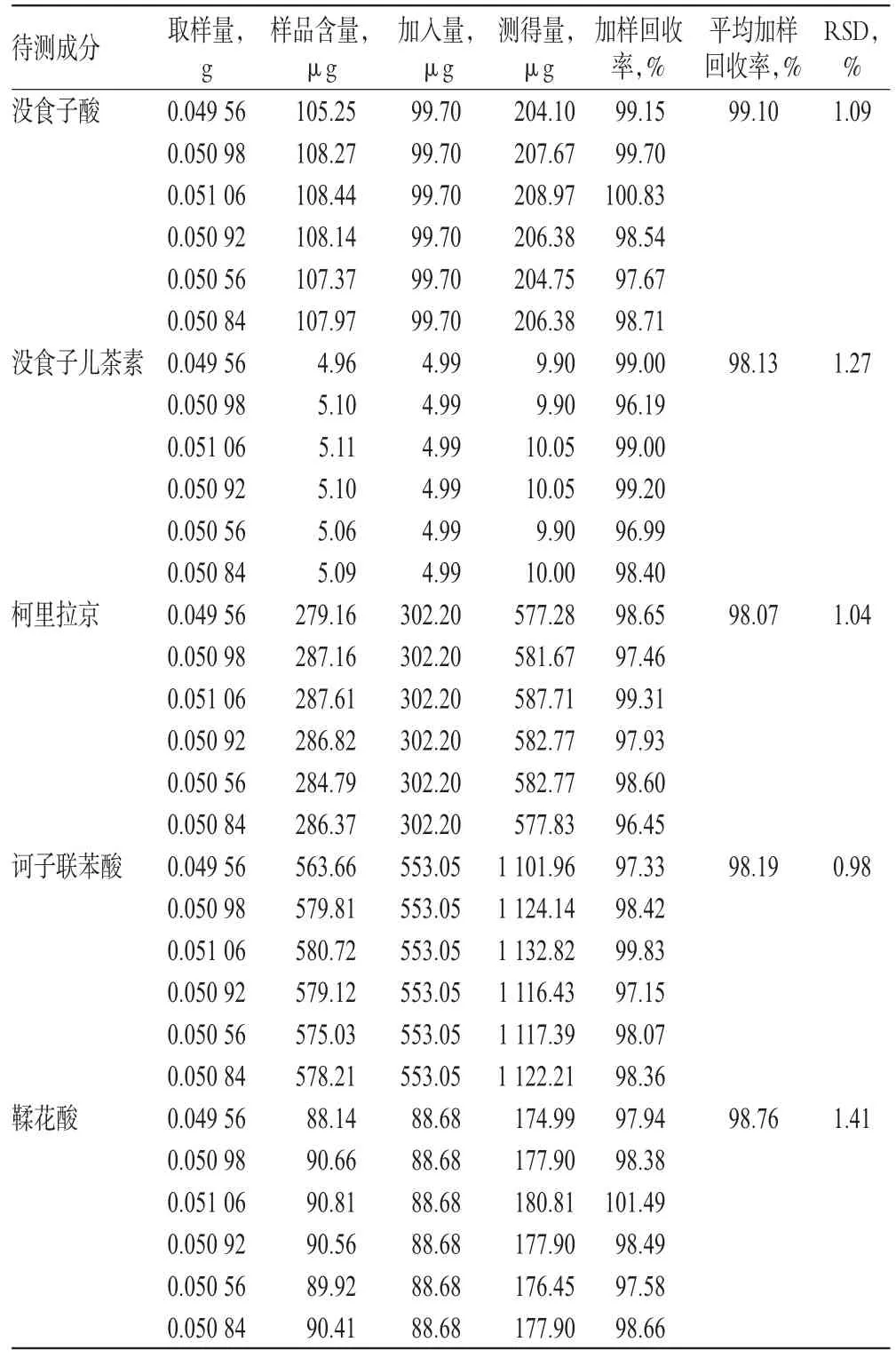
表3 加樣回收率試驗結果(n=6)Tab 3Results of recovery tests(n=6)
2.10 樣品含量測定
各取12批生品約0.1 g,分別按“2.2.3”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并扣除12批生品中含水量后,再計算樣品含量。結果顯示,12批生品中,訶子聯苯酸含量差異最小,其余4種成分含量差異較大,其中沒食子酸含量高低相差近5倍,表明廣西不同產地余甘子藥材質量存在一定差異,結果見表4。
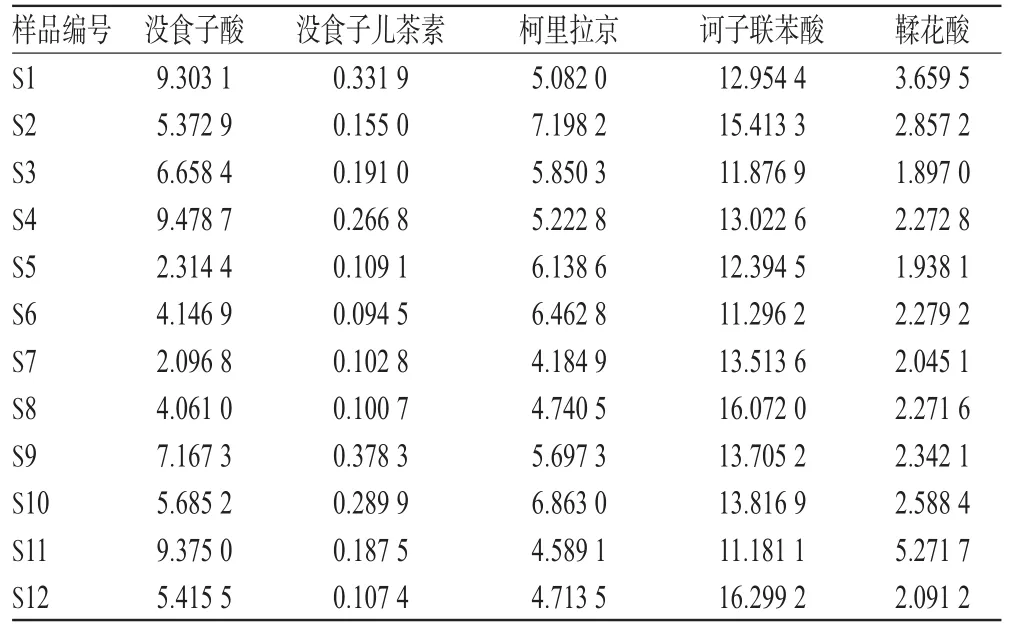
表4 樣品含量測定結果(n=3,mg/g)Tab 4 Results of contents determination of samples(n=3,mg/g)
2.11 主成分分析
主成分分析具有降維思想,可以把多個指標轉化為少數幾個綜合指標,使這些少數的綜合指標又能盡量多地反映原來較多指標所反映的信息。通過SPSS 25.0軟件對12批樣品含量測定結果進行主成分分析。結果,特征值>0.95的前3個主成分累積方差貢獻率為85.753%,能基本說明樣本大部分信息,見表5、表6。主成分1貢獻度最顯著,其中沒食子酸、沒食子兒茶素、鞣花酸載荷較高,說明主成分1主要反映了這3種成分指標的信息;主成分2主要體現訶子聯苯酸的信息;對主成分3貢獻最大的是柯里拉京。根據各主成分得分進行綜合評價,其綜合得分(F)=0.513 1×F1+0.265 4×F2+0.221 5×F3,結果發現所收集的樣品中編號S9的綜合得分(F)最高,藥材質量較好,結果見表7。
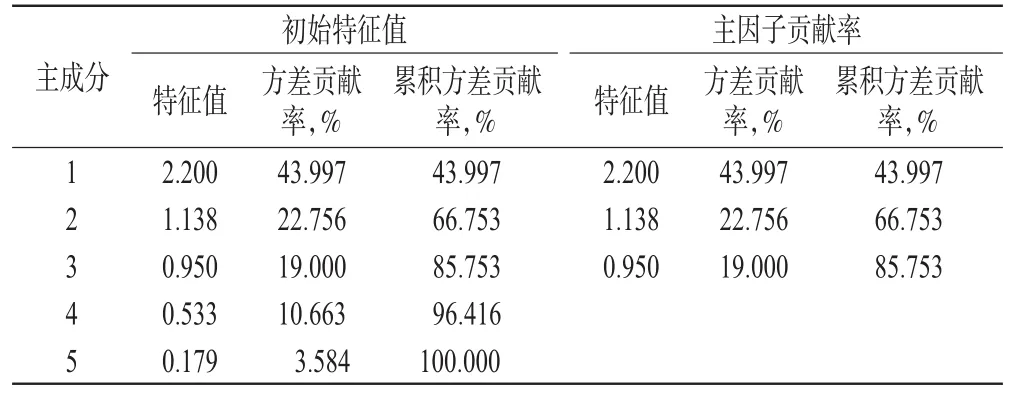
表5 主成分特征值和方差貢獻率Tab 5 Characteristic value and variance contribution rate of principal component
2.12 聚類分析
為了考察廣西余甘子中5種成分的含量差異與產地之間的相互關系,通過SPSS 25.0軟件和歐式距離系數對12批樣品進行聚類分析。當15<閾值<25時,可分為兩類,第Ⅰ類:S11;第Ⅱ類:S1、S2、S3、S4、S5、S6、S7、S8、S9、S10、S12。結合主成分得分排序,可知S11質量稍差。當5<閾值<9時,可將第Ⅱ類細分為四類,第1類:S4、S9、S1,在表7評價排序中分別為第4、1、3位;第2類:S2、S10,在表7評價排序中分別為第5、2位,第1類和第2類又可聚成一類,說明這兩類質量較好,可見排第1、2位廣西玉林市容縣的質量最好。第3、4類中S12、S3、S8、S5、S6、S7在表7評價排序中為第6~12位(第9位除外),說明這兩類質量稍差,結果見圖2。
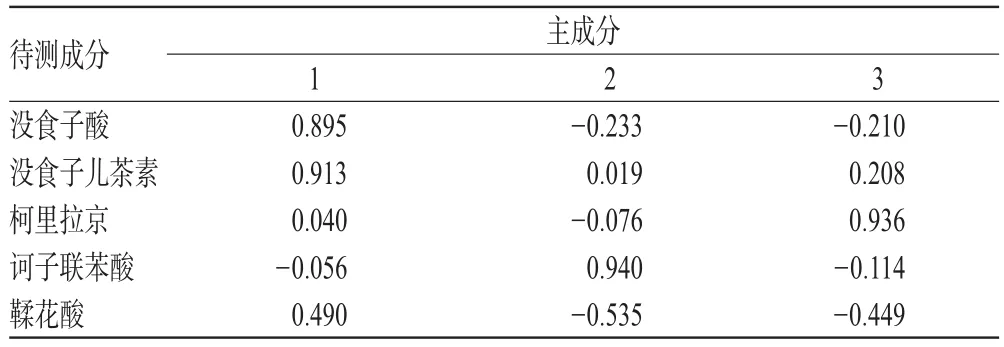
表6 主成分矩陣Tab 6 Principal component matrix
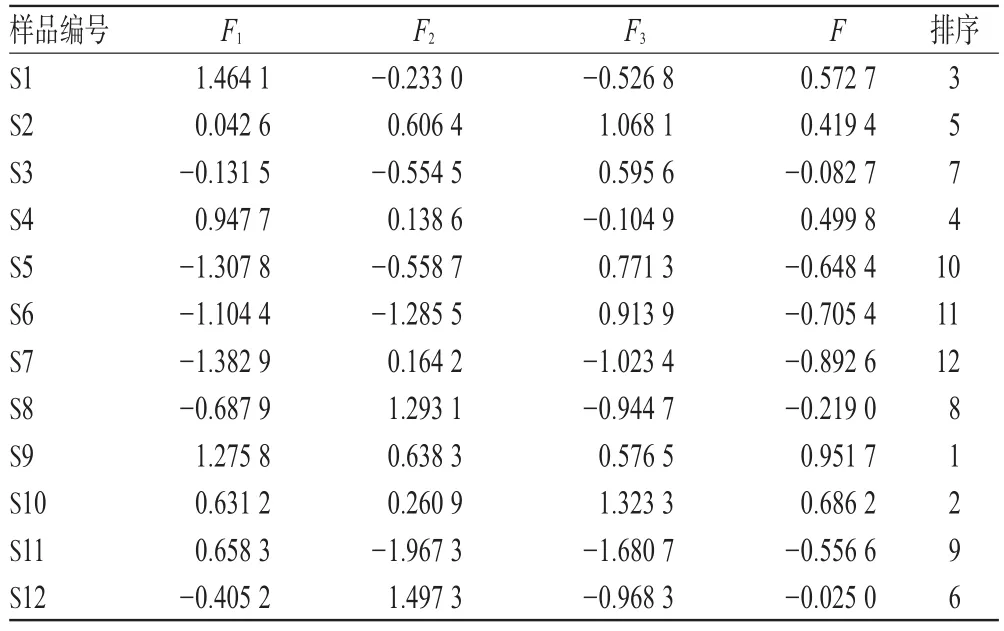
表7 主成分得分綜合評價結果Tab 7 Results of comprehensive principal component evaluation
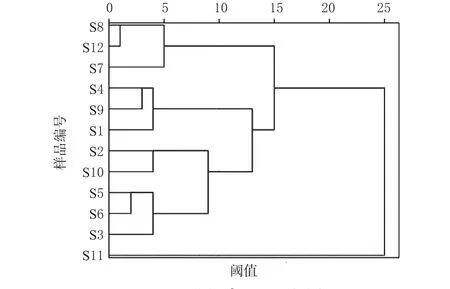
圖2 聚類分析樹狀圖Fig 2 Cluster analysis tree diagram
3 討論
筆者在預試驗中考察了用25%甲醇、50%甲醇、甲醇對余甘子的超聲提取率,結果其提取率分別為11%、19%、19%,結果顯示50%甲醇和甲醇的超聲提取率較高。因鞣花酸具水難溶性[15],在低濃度甲醇中溶解性能較差,而高濃度甲醇提取出的雜質成分較多,檢測易造成干擾,故采用50%甲醇作為提取溶劑。預試驗二極管陣列檢測器圖譜分析結果顯示,沒食子酸、沒食子兒茶素、柯里拉京、訶子聯苯酸和鞣花酸分別在270、220、280、254 nm波長處有最大吸收。在220 nm波長處基線平穩,色譜峰相互之間分離效果較好,且各峰形較好,故選擇檢測波長為220 nm。當有機相選用甲醇時,流動相梯度洗脫過程中基線波動較大,而當乙腈-0.1%磷酸水溶液為流動相時,基線平穩,各色譜峰分離效果也較好,故選擇乙腈-0.1%磷酸水溶液為流動相。
在化學結構上,多元酚類化合物是氫或電子供體,因具有酚類游離基中間體的共振非定域作用和沒有適合分子氧進攻的位置,所以比較穩定,不會引起新的游離基或者因鏈反應而被迅速氧化[16],因此具有抗氧化作用。另據文獻[17]報道,多元酚類化合物在余甘子中含量高達12.9%,而沒食子酸是余甘子中多元酚類化合物的主要成分。這與表6中沒食子酸、沒食子兒茶素在主成分1中載荷較高相一致。由表4可知,沒食子酸含量在廣西不同產地之間差異較大,梧州市藤縣和平鎮的沒食子酸含量為9.478 7 mg/g,而欽州市靈山縣平南鎮的沒食子酸含量為2.096 8 mg/g,相差近5倍,說明產地不同,化學成分的含量存在較大差異,為確保余甘子藥材質量,應相對固定產地采收。經主成分分析,表7中F值越大說明余甘子中成分綜合含量越高,質量越好,由此可知S9質量較好。經聚類分析,廣西不同產地余甘子質量有差異。基本按地域性聚成2類,其中1類又可細分為四小類:玉林市容縣、梧州市藤縣等地區分別聚成質量較好的兩小類,崇左市龍州縣、欽州市靈山縣及百色市田林縣等地區分別聚成質量稍差的另兩小類,該分類符合地域分布特征。但梧州市藤縣個別地區與崇左市龍州縣地區聚成一類,這也說明除不同分布區地理和氣候因子影響外[18],造成同一分布區質量差異還可能與當地的種植栽培條件等因素有關。
綜上,本研究建立的方法操作簡便、結果準確可靠,可用于余甘子中沒食子酸、沒食子兒茶素、柯里拉京、訶子聯苯酸、鞣花酸含量的同時測定。另外,在同時測定余甘子中5種成分含量的基礎上,結合主成分分析和聚類分析,提示廣西不同產地余甘子質量有差異。
[1]國家藥典委員會.中華人民共和國藥典:一部[S].2015年版.北京:中國醫藥科技出版社,2015:179.
[2]南京中醫藥大學.中藥大辭典[M].2版.上海:上海科學技術出版社,2014:1387-1388.
[3]亓旗,崔雅萍,梁文儀,等.藏藥余甘子與訶子化學和藥理作用比較[J].世界科學技術(中醫藥現代化),2016,18(7):1171-1176.
[4]姜紅,史亞軍,趙生玉,等.基于偏最小二乘法對三果湯抗氧化作用譜-效關系的分析[J].中國實驗方劑學雜志,2018,24(3):8-12.
[5]沙磊,張蘭珍,徐義俠,等.RP-HPLC法測定余甘子中訶黎勒酸、沒食子酸、粘酸-2-O-沒食子酸酯的含量[J].藥物分析雜志,2011,31(2):293-295.
[6]張鴻雁,沙磊,石任兵,等.RP-HPLC法測定不同產地余甘子藥材和鞣質有效部位中3種成分的含量[J].中華中醫藥雜志,2012,27(11):2834-2838.
[7]朱智蕓,趙毅,鄧林,等.UPLC法測定余甘子中4種抗氧化活性成分的含量[J].中藥材,2017,40(12):2894-2897.
[8]周利兵.統計學方法中的多元分析和灰色系統理論在化學中的應用研究[M].北京:北京理工大學出版社,2017:132-146.
[9]孫雪飛,張鴻雁,夏青,等.藏藥余甘子藥材及其鞣質部位的指紋圖譜評價研究[J].中國中藥雜志,2014,39(7):1173-1178.
[10]軒轅歡,魏敏,田紅林,等.HPLC法測定余甘子不同提取部位中沒食子酸的含量[J].中國藥房,2015,26(33):4743-4745.
[11]王元清,韓彬,向榮,等.總量統計矩結合聚類分析與主成分分析評價虎杖飲片一致性與差異性[J].中草藥,2015,46(19):2863-2869.
[12]葛重宇,龐慧,李楠,等.18家企業阿膠中氨基酸的含量分析與比較研究[J].中國藥房,2017,28(1):122-126.
[13]莢麗麗,魏嵐,趙健,等.了哥王藥材中7種成分的含量測定及其主成分、聚類分析[J].中國藥房,2017,28(33):4706-4710.
[14]徐柯心,尹澤楠,張文婷,等.雞骨草UPLC指紋圖譜研究[J].藥物分析雜志,2018,38(1):168-174.
[15]武花花.包埋技術在天然產物中的應用研究[D].北京:北京化工大學,2010.
[16]彭知云.沒食子兒茶素電子等排體的合成及抑制尿素酶活性研究[D].吉首:吉首大學,2013.
[17]KUMAR GS,NAYAKA H,DHARMESH SM,et al.Free and bound phenolic antioxidants in amla(Emblica officinalis)and turmeric(Curcuma longa)[J].J Food Compos Anal,2006,19(5):446-452.
[18]熊儀俊,姚小華,王開良,等.我國余甘子地理氣候分類及其特征分析[J].江西農業大學學報,2003,25(2):215-221.
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
