5例性發育異常合并異位腎上腺組織的臨床分析
2018-07-20 09:14:06史精華黃禾鐘定榮陳博陳蓉田秦杰
生殖醫學雜志 2018年7期
史精華,黃禾,鐘定榮,陳博,陳蓉,田秦杰
(中國醫學科學院 北京協和醫學院 北京協和醫院婦產科,北京 100730)
性發育異常是一類因性染色體、性腺或性激素異常而性別表現不典型的先天性異常,雖為少見病,卻對患者本人及家庭造成嚴重的身心壓力[1]。對于含有Y染色體而社會性別為女性的患者有發育不良睪丸容易發生腫瘤或惡變,通常選擇性腺切除。其性腺病理多表現為發育不良或合并發生腫瘤[2]。性發育異常的患者其性腺組織發現異位腎上腺組織罕見,國內外鮮有報道。我們對2007年8月至2017年8月收治的139例性發育異常患者的臨床資料進行回顧分析,發現有5例合并異位腎上腺組織,現報道如下。
臨床資料與結果
病例1 患者女,23歲,因附件區包塊于2014年8月入院。既往因“身高低于同齡兒”于9歲就診我院內分泌門診。查體發現發際低,內眥贅皮,肘外翻,染色體提示:45,X,診為Turner綜合征。予生長激素治療8年后身高為158 cm,一直無月經來潮,以后轉婦科內分泌門診就診,查乳腺發育I級,陰毛III級,口服結合雌激素(倍美力,惠氏制藥有限公司,美國)0.625 mg qd,第15天開始加用甲羥孕酮(安宮黃體酮,上海信誼)4 mg×14 d,無月經來潮,檢查骨齡,骨骺未愈合,行利維愛1.25 mg qd治療,骨骺愈合后改為戊酸雌二醇片/雌二醇環丙孕酮片復合片(克齡蒙,先靈藥業,德國)1片qd,可有撤退出血。2011~2014年周期性使用雌二醇片/雌二醇地屈孕酮片復合片(芬嗎通,蘇威制藥,荷蘭),月經為5~6/28 d;用藥期間監測乳腺及盆腔B超,2013年7月23日B超:右附件區見中高回聲,范圍約1.7 cm×1.8 cm;無自覺癥狀,腫瘤標記物陰性;隨診中發現包塊增大、查人類性別決定基因(SRY)(+)。于2014年8月15日于我院行腹腔鏡雙側附件切除術,術后病理:(右側性腺)惡性腫瘤,結合形態及免疫組化,符合無性細胞瘤;(左側性腺)少許腎上腺皮質結構及散在午菲氏管殘余,卵管未見特殊。術后予以順帕+博萊霉素+依托泊苷(PEB)化療兩程,術后1年應用芬嗎通規律撤退出血;隨訪36個月,子宮4.3 cm×3.7 cm×2.7 cm,月經周期規律,腫瘤無復發,余無特殊。
病例2 患者社會性別女,8歲,因有家族17α羥化酶缺乏病史,該患兒查染色體為46,XY,遂就診我院內分泌科及婦科內分泌。監測血壓140~150/80~90 mmHg;化驗結果顯示:促腎上腺皮質激素(ACTH)28.19 pmol/L,皮質醇23.17 nmol/L,24 h尿皮質醇(urine free cortisol,UFC)0 nmol/24 h,醛固酮(aldosterone,ALDO) 0.47 nmol/L(臥位),血漿腎素活性(Plasma renin activity,PRA) 0.45 μIU/ml(立位),17α羥孕酮(17a-OHP)1.60 nmol/L,孕酮高,睪酮低,高血壓,低血鉀;臨床表現無第二性征發育,陰道呈盲端;B超未見子宮及卵巢,CT雙側腎上腺結節樣增粗,考慮17α羥化酶缺乏。2015年7月腹股溝區超聲:雙側腹股溝區低回聲,不除外睪丸,為行腹腔鏡性腺切除入院。入院查體身高150 cm,體重52 kg;外陰幼稚女性型,無陰毛,陰蒂不大,陰道與尿道分別獨立開口,陰道深約4 cm。術前調整至血鉀正常,術中未見子宮及輸卵管,附件區各可見直徑1.5 cm大小性腺,切除病理提示:(左右性腺)發育不良的睪丸組織,未見附睪;其中(右側性腺)可見少量異位的腎上腺組織。術后復查:ACTH 6.78 pmol/L,ALDO 0.19 nmol/L(臥位),PRA 0.11 μIU/ml(立位);予以潑尼松 5 mg qd,隨診15個月,骨齡與年齡大致相當;激素水平LH 27.03 U/L,FSH 89.96 U/L,E2<19 pmol/L,P 15.42 nmol/L,T<0.35 nmol/L,因年齡小,暫時未用性激素補充治療。
病例3 患者社會性別女,13歲,2017年1月上感惡心嘔吐,伴四肢搐搦、強直、乏力,可自行緩解,同時出現煩渴、多飲多尿,當地測血壓正常(具體不詳),血鉀 1.7 mmol/L,考慮“低鉀血癥”。予枸櫞酸鉀口服后體溫恢復正常,惡心嘔吐、四肢搐搦好轉,停止補鉀后再次出現惡心嘔吐。1月23日轉診至市中心醫院,查血鉀低、尿鉀升高,ACTH 52.85 pmol/L,皮質醇<13.79 nmol/L,24 UFC 103.22 nmol/24 h,ALDO 0.23 nmol/L(臥位),PRA 0.11 μU/ml(立位);FSH和LH升高、雌雄激素水平低、孕激素水平增高;影像學提示雙腎上腺增粗,盆腔超聲未見子宮或卵巢,核型為46,XY;左手腕骨骨齡片:相當于4歲女孩(實際年齡13歲);兒時有腹股溝包塊手術史;經腹婦科超聲:未見子宮、卵巢結構;雙側腹股溝超聲:未見包塊;查體:身高 150.5 cm,體重 39 kg,血壓100/73 mmHg,全身膚色較深,雙乳I期,幼女型外陰,陰毛I期,腹部、腹股溝未觸及包塊。考慮先天性腎上腺皮質增生(congenital adrenal hyperplasia,CAH),予氫化可的松早5 mg、晚7.5 mg和補鉀治療,用藥后上述癥狀逐漸好轉,每周監測血鉀水平正常。7月12日考慮“CAH、17α羥化酶缺乏”收入我院。術前予以氫化可的松早5 mg,中5 mg,晚10 mg,腹腔鏡探查見雙附件區附睪及睪丸樣結構2 cm×3 cm大小,無子宮;切除性腺病理回報(左右性腺)發育不良的睪丸組織,其中(右性腺)可見少許腎上腺皮質組織。術后予以戊酸雌二醇(補佳樂,拜耳醫藥,德國)1 mg qd+氫化可的松 5 mg tid,輔以補鈣,復查ACTH 37.44 pmol/L,ALDO 0.23 nmol/L(臥位),PRA 0.11 μU/ml(立位),隨診3個月無特殊。
病例4 患者社會性別女,12歲,因“出生后陰蒂增大,體毛增多3年”就診于我院。自出生即陰蒂大小約1.0 cm×0.5 cm,逐漸增長。2010年(6歲)外院查染色體46,XY;查體雙側腹股溝似可觸及0.5 cm×0.5 cm大小腫物,可活動,大陰唇發育好,小陰唇發育差,陰蒂肥大,長約2.5 cm,似陰莖,可見正常尿道外口及陰道外口;盆腔B超提示可見片狀子宮11 mm×5 mm×4 mm,未見明顯卵巢,雙側腹股溝區未探及明確睪丸樣回聲;遂行腹腔鏡探查+疝囊高位結扎+肥大陰蒂切除術,術中可見發育不良子宮及“雙側卵巢”,“右卵巢”發育差,與輸卵管傘分離,雙側內環口未閉,內未探及睪丸及輸精管,予以環扎;保留陰蒂頭切除陰蒂體直至海綿體腳,陰蒂切除后送檢病理示符合陰蒂組織學改變;術后患者陰蒂繼續增大,體毛漸增多(胡須、腿毛),皮膚粗糙,音色改變。遂于2016年(12歲)就診我院,查體:身高156 cm,體重53 kg,雙乳房1級無腋毛,大陰唇著色有皺褶,陰蒂長約2 cm,陰毛女性分布,4級;骨齡相當于11~12歲;ACTH 6.98 pmol/L,皮質醇 427.59 nmol/L,甲功、空腹胰島素、AFP、CEA、GH、17α-OHP 均正常,胰島素樣生長因子1(insulin-like growth factor1,IGF1)升高 78.05 nmol/L;FSH、LH及 T 明顯升高;2016年1月超聲:盆腔見子宮大小1.9 cm×0.8 cm×1.5 cm,未及確切卵巢回聲,右側腎上腺前支走形稍迂曲欠均;Y染色體微缺失基因檢測:沒有無精子因子(AZF)缺失;腹腔鏡探查見盆腔偏左幼稚子宮,左側可疑條索樣性腺,右側小睪丸樣性腺直徑約2 cm。行雙側性腺切除+保留血管神經的陰蒂縮短術,術后病理:(左側性腺)輸卵管組織,系膜可見小灶腎上腺組織;(右側性腺)輸卵管組織及發育不良的睪丸組織;(海綿體)符合海綿體組織。術后診斷部分型46,XY性腺發育不全,予以雌二醇片/雌二醇地屈孕酮片復合片治療,3個月后月經初潮,隨診半年無不適。
病例5 患者社會性別女,13歲,因“間斷頭暈、乏力3年,發現血壓升高8月”就診于我院內分泌科。10歲起無明顯誘因出現間斷頭暈、乏力;12歲6個月查體發現血壓升高150~170/110~120 mmHg,血鉀3.25 mmol/L,肝腎功無異常,尿蛋白(±),24 h尿蛋白 0.4 g;ACTH 28.76 pmol/L,皮質醇14.07 nmol/L,24 UFC低于可測值,ALDO 0.66 nmol/L臥位),PRA 0.34 μU/mL(立位);FSH和LH升高、雌激素水平低;染色體核型46,XY;基因檢測提示CYP17A1純合突變;給予卡托普利(開博通,百時美施貴寶,美國)12.5 mg tid、螺內酯(杭州民生藥業)20 mg qd、補達秀(廣州邁特興華制藥)0.5 g tid降壓補鉀,氫化可的松早10 mg、晚20 mg治療,控制欠佳入院。入院查體:身高162 cm體重56 kg,體重指數(BMI)23.01 kg/m2;外陰幼女型,陰道與尿道口分開,可見陰道開口,無陰蒂肥大,陰毛I期;雙側腹股溝未觸及包塊;雙乳I期,骨齡相當于8歲(實際年齡13歲);既往:母親孕期曾服“轉胎丸”;1歲半時因“腸套疊”行手術,5歲時發現右腹股溝腫物,當地醫院考慮“腹股溝疝”行手術治療。入院后改用地塞米松0.75 mg qn,并轉入婦產科,術中見左側睪丸與附睪位于腹股溝管內口處,右側性腺粘連于盆壁,子宮缺如。術后病理回報:(左右)發育不良的睪丸組織,另見異位的腎上腺組織。術后地塞米松減至0.375 mg qn,血鉀正常。術后予以補佳樂1 mg qd補充治療促進發育。
5例患者術前血清性激素水平測定結果見表1。
表15例患者術前血清性激素水平測定結果(為早卵泡期參考范圍)
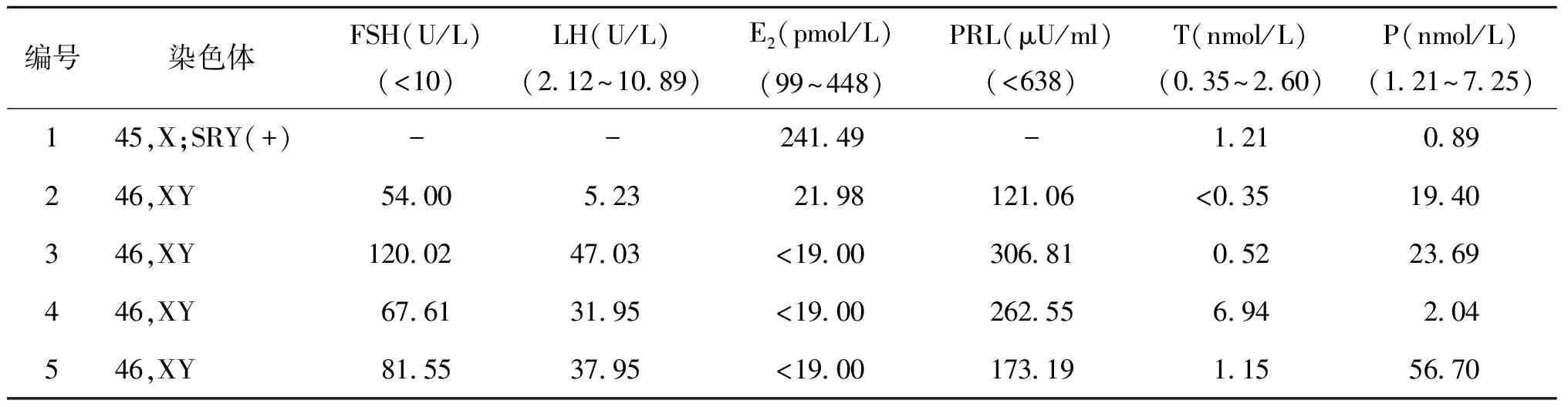
編號染色體FSH(U/L)(<10)LH(U/L)(2.12~10.89)E2(pmol/L)(99~448)PRL(μU/ml)(<638)T(nmol/L)(0.35~2.60)P(nmol/L)(1.21~7.25)145,X;SRY(+)--241.49-1.210.89246,XY54.005.2321.98121.06<0.3519.40346,XY120.0247.03<19.00306.810.5223.69446,XY67.6131.95<19.00262.556.942.04546,XY81.5537.95<19.00173.191.1556.70
表25例患者術后血清性激素水平測定結果
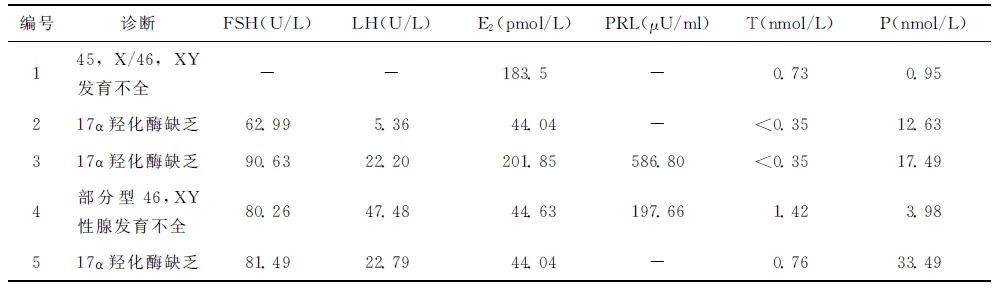
編號診斷FSH(U/L)LH(U/L)E2(pmol/L)PRL(μU/ml)T(nmol/L)P(nmol/L)145,X/46,XY發育不全--183.5-0.730.95217α羥化酶缺乏62.995.3644.04-<0.3512.63317α羥化酶缺乏90.6322.20201.85586.80<0.3517.494部分型46,XY性腺發育不全80.2647.4844.63197.661.423.98517α羥化酶缺乏81.4922.7944.04-0.7633.49
注:“-”表示未查
附:發育不良睪丸合并異位腎上腺皮質病理圖

大部分曲細精管都只見支持細胞,僅罕見間質細胞,并且管腔大部分未打開,少數打開的管腔中僅極少數含有生精細胞,且生精細胞并不處在發育的各個階段圖1 發育不良的睪丸組織(HE染色 ×100)
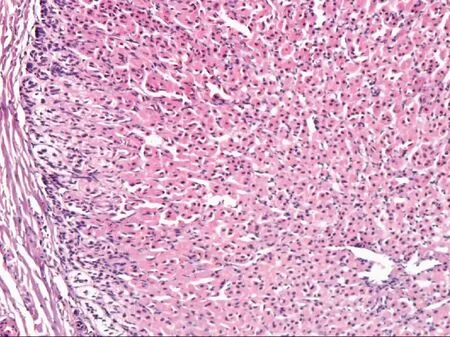
可見腎上腺皮質結構,球狀帶、束狀帶、網狀帶結構依次存在,細胞形態和排列,與正常的腎上腺皮質一致,未見腎上腺髓質結構圖2 異位的腎上腺皮質(HE染色 ×100)
討 論
一、異位的腎上腺組織
異位腎上腺組織并非罕見,主要見于靠近腎上腺的腹膜后,也較常見于腎臟、睪丸、精索、附睪及陰囊。此外,在顱腦、脊柱[3]、肝膽胰[4]、胎盤、闌尾、橫結腸、闊韌帶、卵巢[5]、腹膜、疝囊、肺部和心包膜等部位也有報道。多無功能且無明顯癥狀,于手術治療時才在手術部位意外發現[6]。無病變的異位腎上腺組織表現為黃色圓形或卵圓形結節,與周圍組織界限清晰,直徑1~5 mm不等。病理上應與發育不良的曲細精管、透明細胞轉移腫瘤、脂肪母細胞瘤等相鑒別。一旦發現應予以完整切除,但無需術中常規探查[7]。
腎上腺組織異位于性腺多見于小兒泌尿外科手術,有報道至少50%的新生兒和嬰兒可發現異位腎上腺組織。Dobanovacki等[8]報道了3 028例1~17歲患兒行腹股溝手術,術中及術后發現異位腎上腺組織在隱睪、鞘膜積液和腹股溝疝,發生率分別為2.4%、2.3%和2.0%。但婦科報道罕見。由于正常腎上腺的存在,隨著年齡的增長絕大多數異位腎上腺組織將發生萎縮直至消失,只有少數得以殘留,又被稱為副腎上腺組織或腎上腺殘余。異位腎上腺大多數只有皮質成分,少數既有皮質又有髓質。同原位腎上腺一樣有增生或腫瘤的可能,因而如術中發現異位的腎上腺成分應同時切除[9]。
究其腎上腺組織異位原因,目前較為認可的為胚胎缺陷學說[10]:腎上腺發生于生殖腺附近,由共同原基形成。其皮質由位于后腹壁腸系膜根部與尿生殖嵴之間的間皮細胞群發生而來,而髓質由交感神經嵴衍生而來。隨著髓質細胞向皮質區域遷移,腎上腺組織的碎片,尤其是腎上腺皮質的碎片可能被分裂開來。大多數碎片停留在正常腎上腺附近,少數與尿生殖嵴關系較為緊密的碎片則可能隨著性腺遷移而異位。器官培養研究[11]表明,ACTH反應性細胞存在于生殖腺/中腎的邊界,同時腎上腺類固醇細胞可以轉移到XY性腺,我們的報道也與其相符,5例異位患者,4例染色體為46,XY,剩余一例雖未見Y染色體但SRY基因陽性,合并有無性細胞瘤。MacMahon等[12]報道了一例45,X/47,XY,+18染色體嵌合型的性腺發育不全患者考慮雙側性腺腫瘤,術中切除性腺的病理提示有異位的腎上腺組織,但文章并未解釋這一現象。
二、先天性腎上腺皮質增生對異位腎上腺組織的影響
CAH是一種腎上腺激素合成紊亂的遺傳性疾病。CAH合并睪丸腎上腺殘余腫瘤(Testicular adrenal rest tumour,TART),又稱為腎上腺生殖綜合癥,自從1940年報道以來陸續增多,發病率差異大,介于0~94%之間[13]。正常兒童性腺內腎上腺殘余組織約1歲左右自然退化,CAH患者在控制不佳時,ACTH和血管緊張素Ⅱ增高,可能會刺激這些殘余細胞增生,發生腎上腺殘余瘤。年齡越大,CAH控制越差,TART發病率越高[11]。本文中一半以上(3/5)的患者均是由17a-羥化酶缺乏所致CAH。分析垂體代償性分泌ACTH引起異位和在位腎上腺皮質增生,阻礙了睪丸內異位殘余腎上腺細胞的退化,故而在兒童期仍然發現有殘存的腎上腺組織。兩例患者手術時均為兒童,如果未能及早發現并控制CAH,待成人后很可能會進展為TART。CAH合并TART治療的經驗表明,術中如發現異位的腎上腺組織應予以切除(尤其是腫瘤形成),對于術后治療CAH有顯著的改善作用。
同為性腺組織,然而卵巢腎上腺殘余腫瘤(ovarian adrenal rest tumor,OART)發病卻很罕見,至今國內外均是散在的個案報道[14]。Boyer等[15]認為Wnt4可能是TART及OART發病率差別的原因之一。女性性腺發育過程中需要Wnt4阻止合成類固醇細胞從中腎遷移至發育中的卵巢,抑制CYPl7A1(Cytochrome P450 17A1,細胞色素17A1)及HSB3B2酶(type 2 gene of 3β-hydroxysteroid dehydrogenases,3β羥類固醇脫氫酶2型)的活性,抑制異位類固醇合成。因而對腎上腺原基細胞發育為OART或遷移至卵巢性腺均有抑制作用,僅當女性患者在ACTH的刺激下,且Wnt4的抑制作用不足才可發生OART,因而發病率低。此外,因為很少在年輕的女性患者中切除不發育的性腺,因此發現OART的機會更低。
關于性發育異常患者合并異位腎上腺組織國內尚缺少相關報道,我們對其發生的可能機制和治療的原則多借鑒于睪丸異位腎上腺組織的情況。文獻的回顧結合我們報道的5例患者,我們認為對于性發育異常尤其合并有CAH的患者,臨床醫生和病理醫生應提高異位腎上腺組織的意識和加強對其診治,一旦發現應予以切除并嚴密隨診。隨著對該疾病的發現,也期待有更多的病例資料的報道,以總結經驗提高對該疾病診治的認識。
