顱骨上皮樣血管肉瘤1例及相關文獻復習
2018-07-27 12:10:10毛椿平
山西醫科大學學報 2018年7期
關鍵詞:信號
周 榮,毛椿平*,饒 蘭
(1荊門市第一人民醫院放射科,荊門 448000;2荊門市第一人民醫院病理科;*通訊作者,E-mail:dr-maochunping@126.com)
上皮樣血管肉瘤(epithelioid angiosarcoma,EA)是一種少見的高度惡性的血管源性腫瘤,多發生于四肢的深部軟組織內,發生于骨骼較為罕見,其在病理組織學上有一定特征性,影像表現缺乏特異性。本例報道發生在顱骨的上皮樣血管肉瘤,旨在提高對本病的認識以及擴展對顱骨占位性病變的診斷思路。
1 病例資料
患者,男,31歲,因“發現右顳部包塊約2月”入院。當地CT提示右顳部占位,侵及顱內及顱外,遂轉入我院。患者一般情況及實驗室檢查無特殊。
頭顱CT平掃示右側顳骨、蝶骨及額骨交界區見一不規則形軟組織密度影,最大截面積約4.3 cm×3.1 cm,邊界清楚,密度不均,病灶周圍可見線樣高密度影,鄰近軟組織及右側眼外直肌受壓(見圖1)。CT診斷:右側顳骨、蝶骨及額骨交界區占位,考慮嗜酸性肉芽腫。
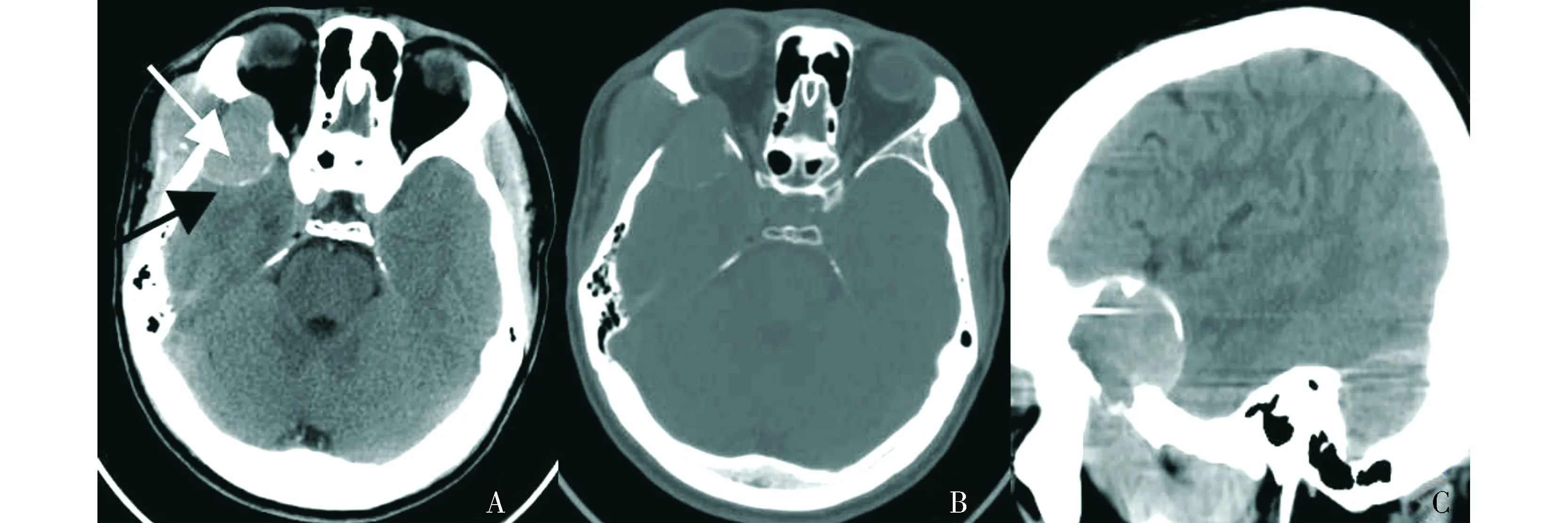
A.橫斷位CT平掃示右側顳骨、蝶骨及額骨交界區不規則軟組織密度腫塊(白箭頭),內部密度不均,邊緣可見線樣高密度(黑箭頭);B.橫斷位CT平掃骨窗示病變區溶骨性骨質破壞,骨皮質變薄呈蟲蝕樣,局部似欠連續;C.MPR矢狀位示病變特征圖1 顱骨上皮樣血管肉瘤患者的橫斷位CT平掃與MPR矢狀位圖像
頭顱MRI平掃示右側顳骨、蝶骨及額骨交界區病灶呈等T1WI混雜T2WI信號(見圖2),DWI可見病灶內散在輕度彌散受限(見圖3),注射Gd-DTPA后增強掃描呈明顯不均勻性強化(見圖4)。MRI診斷:考慮腫瘤性病變。
手術及病理:患者于全麻下經右側翼點入路行右側顳骨、蝶骨及額骨交界區腫塊切除術。術中探查發現腫塊外側與鄰近顳肌部分粘連,界限不清;腫塊內側與硬膜粘連緊密,相應硬膜外層增生;腫塊質軟,血供豐富,呈暗紅色。分塊切除腫塊后送病理檢查。大體標本觀察:6.5 cm×6.0 cm×4.2 cm碎組織一堆,切面呈灰白、灰褐及暗紅色。鏡下見腫瘤細胞片狀分布,瘤細胞為圓形或多邊形的上皮樣細胞,胞核空泡狀,核仁明顯,部分瘤細胞內可見空泡,偶含紅細胞,部分區域可見分支狀血管性腔隙(見圖5)。免疫組化檢查:CD31(+),ERG(+),CD34(部分+),PCK(-),S100(-),INI-1(-),Ki67(LI:5%)。病理診斷:右側顳部(顳骨、蝶骨及額骨交界區)上皮樣血管肉瘤。
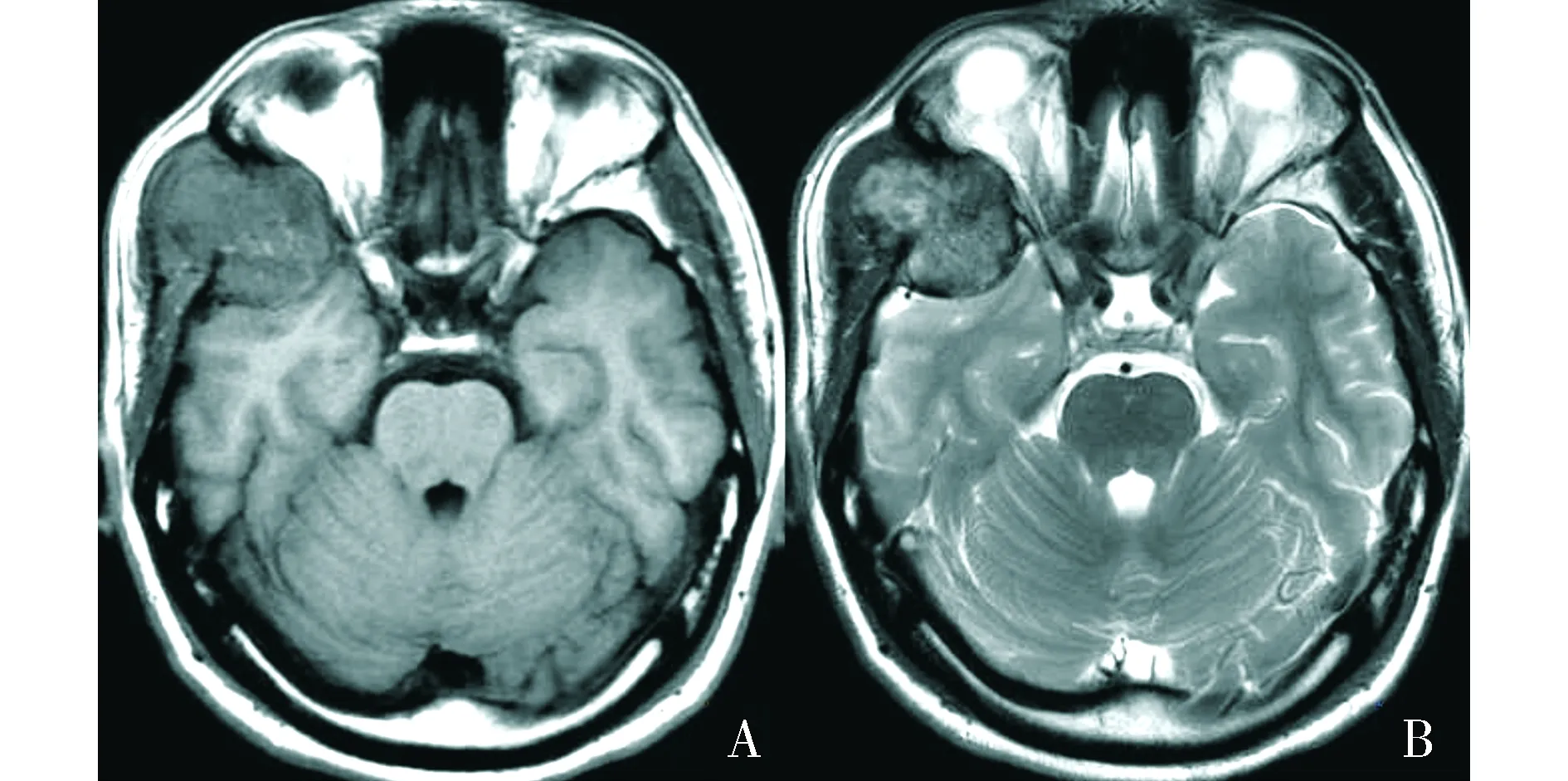
A.橫斷位T1WI示病變呈等信號為主,信號欠均;B.橫斷位T2WI示病變呈稍高信號為主混雜信號圖2 顱骨上皮樣血管肉瘤患者的頭顱MRI平掃影像
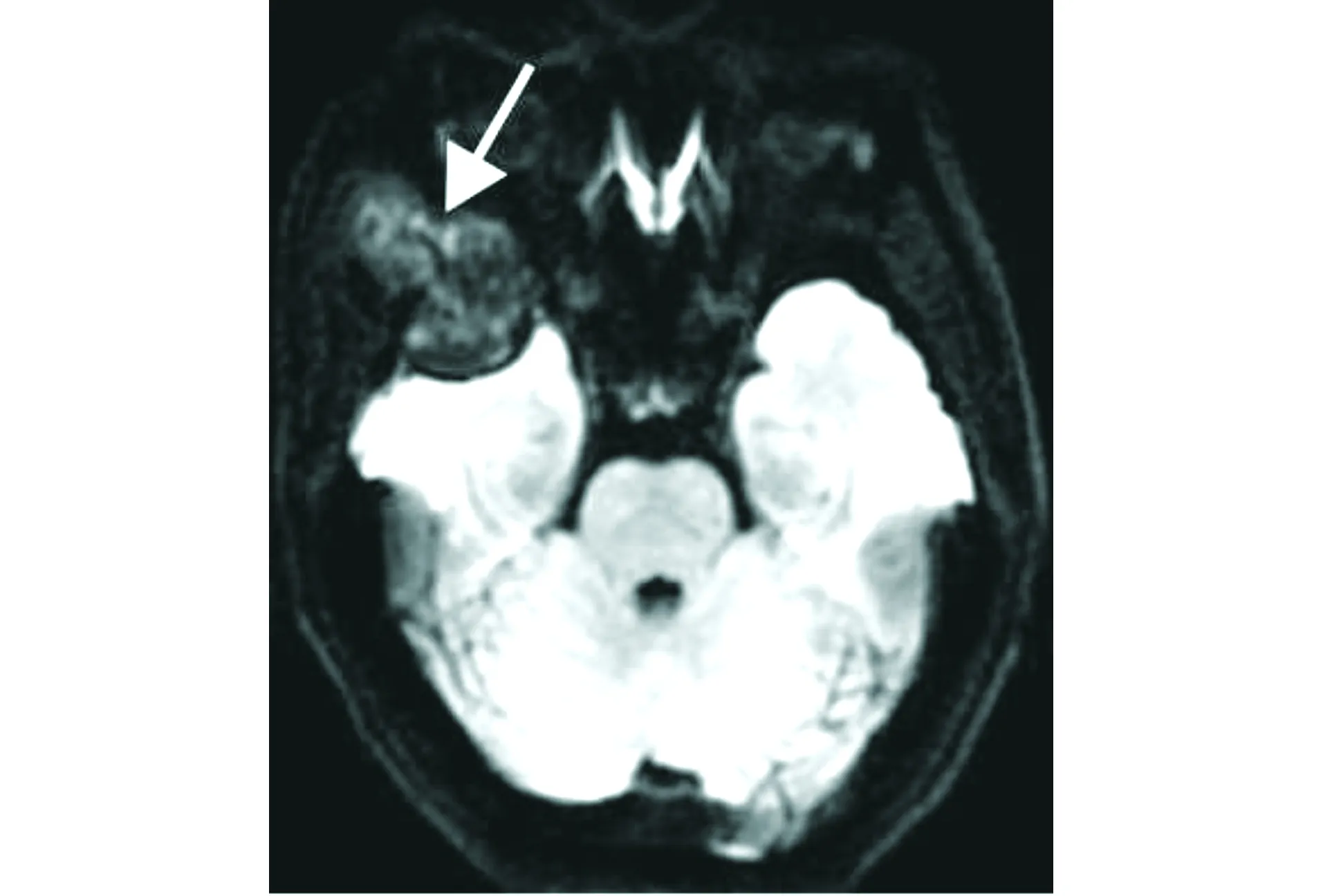
橫斷位DWI示病變內部散在輕度彌散受限改變(白箭頭)圖3 顱骨上皮樣血管肉瘤患者的橫斷位DWI
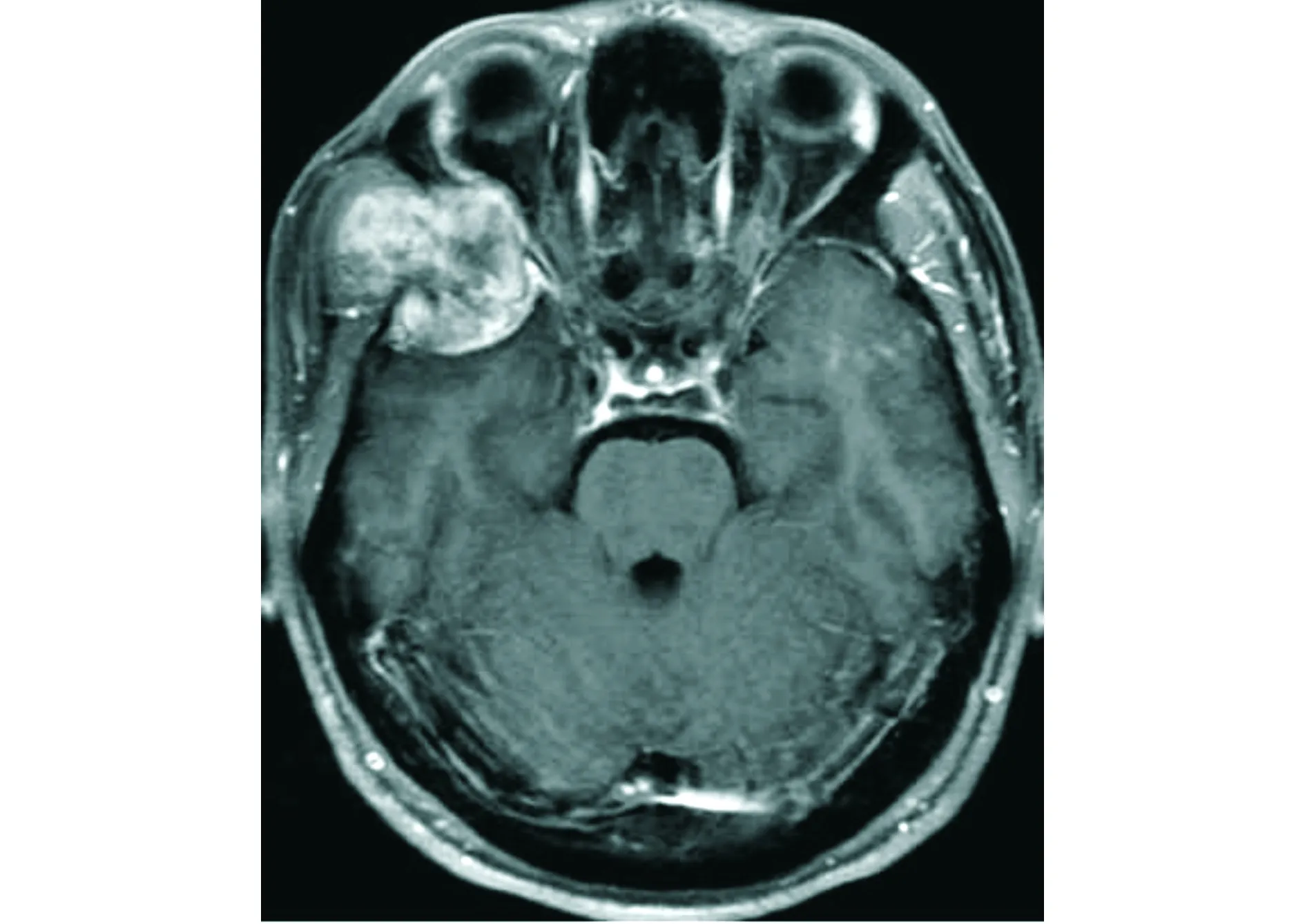
橫斷位T1WI增強示病變呈明顯不均勻性強化圖4 顱骨上皮樣血管肉瘤患者注射Gd-DTPA后增強掃描
2 討論
2.1 概述
上皮樣血管肉瘤于1986年由Perez-Atayde等[1]首次報道,是一種少見的高度惡性的血管源性腫瘤,為血管肉瘤的少見亞型。EA多發生于四肢的深部軟組織內,少見部位包括皮膚、腎上腺、甲狀腺、陰道、膀胱、肺及骨[2]。骨原發性EA較為罕見,僅占骨原發性惡性腫瘤<1%[3],又以下肢骨相對常見,偶見于顱骨、脊椎及骨盆[4]。骨源性EA男性多于女性,以青年人和老年人多見[5],臨床上表現為病變部位的疼痛而無其他特異性癥狀,多以無意發現病變部位包塊就診。本病易沿血管遠處轉移,通常預后較差[6],因此術前準確的影像學診斷對臨床手術及治療價值較大。
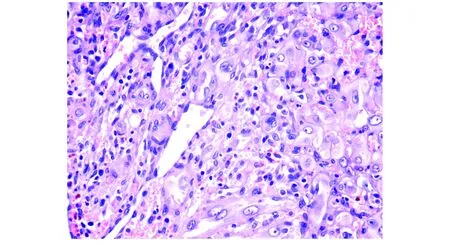
圖5 右側顳部上皮樣血管肉瘤病理表現(HE,×400)
2.2 影像及病理表現
顱骨EA的影像學表現與其組織學惡性程度相一致,同時又缺乏特異性。根據以往報道[7,8]及本例特點,以下幾點CT及MRI表現可能提示本病:①腫塊以顱骨的板障為中心生長,局部膨脹,如浸潤性生長可破壞骨皮質,形成周圍軟組織腫塊;②CT平掃呈不均質軟組織腫塊或溶骨性骨質破壞,其內無鈣化,周邊無硬化邊,一般不伴有骨膜反應;③MRI平掃在T1WI上呈等信號或低信號,在T2WI上呈混雜稍高信號;④增強掃描表現為明顯不均勻性強化。顱骨EA的最終確診仍需依靠病理學檢查,其鏡下表現是腫瘤細胞上皮樣,核膜不規則,核仁明顯,異型性和多形性明顯,壞死、核分裂及病理性核分裂常見。部分細胞內可見胞質內空泡,偶見紅細胞,腫瘤內可見襯異型上皮樣內皮細胞的血管樣腔隙形成。免疫組化表達血管內皮細胞的標志物如CD31、CD34、FactorVIII、FLI-1和ERG[9],彼此聯合應用可提高準確性。
2.3 鑒別診斷
本例顱骨EA表現為顱骨的軟組織腫塊,影像上需與常見的顱骨病變相鑒別。
血管瘤:好發于中老年人,起源于顱骨板障。CT平掃以病灶內放射狀的骨小梁為特征。MRI平掃信號混雜,其內可見條形或點狀低信號。增強掃描,明顯不均勻強化。
孤立性漿細胞瘤:中老年人多見,起源于顱骨板障。CT平掃呈單一的骨質破壞,伴以骨質破壞區為中心的雙凸狀軟組織腫塊。MRI平掃呈等T1WI、稍長T2WI信號,邊緣可見低信號的“假包膜”,無瘤周水腫。增強掃描,明顯強化,無腦膜尾征。
嗜酸性肉芽腫:好發于兒童和青少年,男性多見,起源于顱骨板障,病變一般較小(<3 cm)。CT平掃呈單發或多發溶骨性骨質破壞,呈穿鑿樣,邊緣清晰或伴有不同程度骨質硬化,可見“紐扣樣”死骨,伴較小軟組織腫塊。MRI平掃呈稍長或等T1WI、稍長T2WI信號。增強掃描,輕度至明顯強化,無腦膜尾征。
轉移瘤:常見于50歲以上中老年人,其他部位有原發腫瘤灶。在CT或MRI平掃上呈單發或多發溶骨性骨質破壞,呈蟲蝕樣,可伴有以骨為中心的軟組織腫塊。增強掃描,明顯強化。
間變型腦膜瘤:男性多見,起源于蛛網膜顆粒的帽狀細胞,呈侵襲性生長。好發于上矢狀竇旁、大腦鐮旁及大腦凸面等部位。CT平掃呈以寬基底與顱骨內板相連的等或低密度不規則形腫塊,常有囊變、出血、壞死及鈣化。如伴有溶骨性骨質破壞,可向顱外生長形成梭形軟組織腫塊。MRI平掃呈等或稍長T1WI、稍長T2WI信號,瘤周水腫明顯。增強掃描,明顯不均勻強化,有腦膜尾征。
Rosai-dofoman病:好發于兒童和青少年,男性居多,起源于板障。CT平掃呈溶骨性骨質破壞,伴跨顱骨內外板生長的梭形軟組織腫塊。MRI平掃呈等T1WI、等T2WI信號。增強掃描,明顯均勻強化,腦膜增厚,伸入鄰近腦溝呈“偽足樣”改變。
以上幾種疾病綜合分析,多能明確診斷。本例報道旨在提高對顱骨上皮樣血管肉瘤的認識以及擴展對顱骨占位性病變的診斷思路。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
