土壤樣品中重金屬化學形態模型的發展與應用
2018-08-02 01:46:58鄧迎璇李永濤李曉晶陳雅麗翁莉萍
農業環境科學學報 2018年7期
鄧迎璇,李永濤,2,李曉晶,王 龍,馬 杰,陳雅麗,翁莉萍*
(1.農業部農產品質量安全環境因子控制重點實驗室/農業部環境保護科研監測所,天津 300191;2.華南農業大學資源環境學院,廣州 510642)
土壤樣品中的(類)重金屬元素[如鎘(Cd)、砷(As)]以多種化學形態存在,重金屬在土壤中的生物有效性、生物毒性和遷移性,不但受其總濃度的影響,也取決于其化學形態分布。因此了解和預測土壤樣品中重金屬的化學形態分布,對提高其環境風險評估與管控的準確性與有效性,從而保持生態系統的可持續發展十分重要。
大部分土壤中的重金屬存在于固相中,主要包括:存留于(黏土)礦物顆粒中的雜質、由沉淀作用等形成的難溶解態物質、吸附在各種顆粒表面的吸附態重金屬;液相介質中的重金屬形態主要有:自由態離子、與無機配體結合態、與溶液中的(有機和無機礦物)膠體顆粒物結合的吸附態重金屬。溶液中重金屬的各種形態與其元素在環境中的遷移性密切相關。一般認為,溶液中的自由態離子是能被植物根系直接吸收的化學形態,由此產生了“自由離子活度模型”(Free Ion Activity Model)[1-2]。存在于固相中的吸附態重金屬和部分易于溶解的沉淀態重金屬,由于能較為快速地釋放到溶液中,而被稱為具有反應活性(Reactive)的化學形態。相反,(黏土)礦物結構內部和難以溶解的沉淀中的重金屬,由于短期內不能釋放,而被稱為惰性或沒有反應活性(Non-reactive)的化學形態。由此可見,重金屬的化學形態受吸附、絡合和沉淀等反應過程控制,而吸附反應在控制土壤樣品中重金屬的化學形態分布上起著重要作用。
研究土壤中重金屬化學形態的方法主要有實驗分析和模型計算兩種。土壤樣品中重金屬的全量消解常采用含有氫氟酸的酸溶法和堿熔法,非全量或近全量消解主要采用強酸組合或者強酸與氧化劑的組合。除了土壤全量,重金屬化學形態分析傳統上主要采用具有不同強度和提取機制的提取劑進行振蕩提取[3]。這些提取方法進行土壤重金屬化學形態分析的優勢在于其比較簡便易行,但是存在專一性差、形態解釋與實際不符、提取過程中發生再分配等問題[4]。為了能更好地評估土壤中重金屬的生物有效性,人們又發明了諸如同位素稀釋[5]和梯度擴散薄膜技術(Diffusive Gradients in Thin-films,DGT)等含有動力學意義的形態測定方法[6-7]。僅少數分析方法可以直接測定溶液中重金屬離子形態,如離子選擇電極、(離子交換-)高壓液相色譜、多南膜(Donnan Membrane Technique,DMT)技術等[8-11]。
應用化學分析方法測定元素化學形態具有直接、準確的優勢,但是化學分析方法較為昂貴耗時,且現有的分析方法不能給出一種重金屬在土壤樣品中分布的全貌,常用的分析方法并不具備識別化學形態的特異性。比如:環境中普遍存在的天然有機質(Natural Organic Matter,NOM)能夠結合金屬陽離子如Cd2+,形成NOM絡合態Cd,但是測定這些結合態的金屬含量在方法上難以實施。而且,化學分析方法測得的元素化學形態僅適用于當前測定的樣品,一旦更換研究體系,分析結果很可能將不再適用,因此化學測定分析方法的預測性通常較差。為了克服以上化學分析方法中的缺點,人們開發了利用模型計算與預測元素化學形態的手段。本文綜述了用于土壤環境樣品中重金屬元素化學形態模型的發展,介紹了幾種機理性吸附模型的主要機制,總結了用于預測重金屬離子在天然環境樣品上吸附模型參數的獲取方法,具體模型應用的方法以Cd和As分別作為環境中金屬陽離子和含氧陰離子的代表進行了闡述,并對模型的應用和發展進行了展望,以期為今后化學模型用于元素在天然環境樣品中化學形態分布的預測提供借鑒。
1 經驗性模型與機理性模型
1.1 經驗性模型
環境樣品中重金屬的化學形態模型可分為經驗性模型和機理性模型。經驗性模型是基于實際樣品的實驗室分析數據,通過回歸分析等統計手段建立起來的某種化學形態的濃度或元素分配系數與其他因子的數量關系。對于土壤樣品,此類模型也被稱為土壤傳遞函數(Pedo-transfer function)[12]。
最簡單的經驗性模型是線性(吸附)固-液分配模型:Q=KdC(其中:Q為某種重金屬在固相中的含量,mg·kg-1);Kd為固-液分配系數,L·kg-1;C為該元素在溶液中的濃度,mg·L-1)。一般情況下,線性關系只有在重金屬濃度很低的情況下才成立,而且在不同土壤中Kd值也會隨著重金屬含量、pH、土壤有機質(SOM)含量等因素而變化,其數值變化可能跨越幾個數量級[13-15]。但是為了便于計算,在很多涉及元素固-液分配的生物地球化學模型中,人們仍經常采用單一固定的Kd值,這給模型預測的準確性帶來很大影響。比線性模型略微復雜、適用性更強一些的是Freundlich 模型[16-17]:Q=KFrCn(其中:KFr是吸附/反應常數,其單位取決于參數n的數值;n沒有單位,其數值一般<1,當n=1時Freundlich模型等同于線性模型)。與線性模型相比,Freundlich模型考慮重金屬固-液分配的非線性表現,其適應的重金屬濃度范圍更加廣泛。除了濃度之外,土壤pH等因素也影響到重金屬元素在土壤中的化學形態分布[18]。為了提高模型的普適性,增加了其他因子的擴展,Freundlich模型被廣泛應用。將Freundlich模型進行對數轉換可將模型轉換為線性關系。
類似于Freundlich模型的對數線性經驗性模型被廣泛應用于土壤中重金屬離子(如:Cd2+、Cu2+、Pb2+)化學形態的計算[13,19-20],但針對同種元素不同研究得到的模型考慮的因子有所不同。以Cd為例,在Luo等[19]、Sauve等[13]和 Mcbride等[20]的研究中用于表述土壤溶液中Cd濃度的經驗性模型分別采用了1、2、3個因子作為自變量,公式(1)~公式(3):
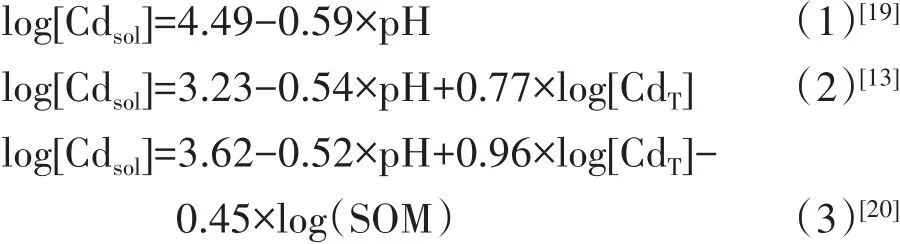
式中:Cdsol為Cd在土壤溶液中的濃度,μg·L-1;CdT為土壤中Cd的總量,mg·kg-1;SOM為土壤有機質含量,%。之所以選擇了不同數量的自變量是受不同作者對模型計算的簡便性和準確性之間權衡的影響,也受到所用數據中不同因子的變化范圍和土壤性狀等其他因素的影響。Luo等[19]研究顯示溶液中的Cd與土壤體系pH呈線性相關[公式(1)],加入SOM并不能顯著提升模型擬合的準確性。另外,即使模型擬合選用相同的自變量,不同文獻中模型參數數值之間通常也存在較大差異。同樣以Cd為例,Sauve等[13]和Hao等[21]都采用了pH和土壤總Cd含量來計算溶液中的Cd濃度,如公式(2)和(4)所示:
log[Cdsol]=-1.515+0.072×pH+0.685×log[CdT](4)[21]
可以看出,同樣是采用pH和CdT這兩個變量因子,但所對應的模型參數相差很大,甚至代表因子影響方向的參數正負符號都可能相反[13,21]。由于模型的經驗性,數據范圍和主要限制因子的變化可能導致擬合的模型參數的不同。
由以上實例可見,經驗性模型具有計算便捷和易于理解掌握等優點,所取得的模型及其參數在一定范圍內能夠很好地表述重金屬離子在固-液介質中的分配,同時也能在一定程度上揭示可能的控制機制。但是,經驗性模型是“黑箱模型”,受模型的統計經驗性制約,模型的使用局限于建模時所采用的參照數據范圍,而土壤體系組分和溶液化學性質的多變性妨礙了這些經驗性模型在不同環境樣品中的應用,致使經驗性模型具有普適性差、機理性不清等缺點。因此經驗模型方法在計算重金屬離子形態上的不足逐漸被人們意識到[13-15,22]。
1.2 機理性模型
土壤中重金屬化學形態的機理性模型是基于對反應過程的機制分析,應用化學反應定律而建立的模型,亦即“白箱模型”。瑞士水化學家Werner Stumm在研究中引入了化學平衡的概念,推動了基礎化學原理在定量計算環境樣品中化學反應過程的應用[23],Stumm相信要深入理解地方、區域和全球性的元素循環和污染影響,分子水平的信息是必不可少的。這一理念也逐漸被土壤化學領域所接受與應用。有關酸-堿反應、配位化學、沉淀-溶解平衡,相轉換過程、氧化-還原過程,表面化學、光化學原理、固體溶解速率和顆粒傳輸等過程的物理化學基本理論與計算方法被逐漸融入到水、土壤等環境介質定量化學模型中。
土壤中重金屬化學形態機理性模型的建立也是基于化學平衡這一概念,基于熱力學原理而構建的。根據土壤的基本組成特點,至少需要考慮:(1)溶液中的化學反應;(2)固-液間的沉淀-溶解過程;(3)溶質在固體表面的吸附-解吸過程。土壤溶液化學反應中大部分過程的計算方法比較成熟,如酸-堿反應采用水解離常數、化合反應采用化合常數、氧化-還原反應根據氧化-還原電位和氧化-還原常數、CO2的溶入應用Henry常數等,所需參數也比較完備。固-液之間的沉淀-溶解反應的計算原理也比較簡單,采用離子溶度積來計算,應用的難點在于沉淀-溶解過程是否達到最終的平衡。沉淀-溶解反應的進程較慢,即使溶質濃度達到超飽和狀態,相應的沉淀也未必形成,或者先形成一種溶解度更高的沉淀、再逐步老化達到最終溶解度低的沉淀物質。土壤中含有多種有重金屬吸附活性的顆粒,主要包括天然有機質、金屬氧化物和黏土礦物。重金屬離子通過化學鍵合(特異性反應)和靜電作用(非特異性反應)吸附在這些顆粒表面,形成多種表面形態。離子的吸附不但受到顆粒表面特征的影響,也取決于pH、離子強度和其他吸附質的存在。如何準確計算離子在溶液中膠體表面和固相中顆粒表面的吸附過程是建立土壤重金屬化學形態機理性模型的主要難點之一。
表面絡合模型(Surface Complexation Model,SCM)是用于表征和計算離子等吸附質在吸附劑顆粒表面結合與富集過程的物理化學模型。常用的 Langmuir[24-25]模型[公式(5)]反映了表面位點的概念和位點飽和現象:

式中:KLa為吸附常數,L·kg-1;C為平衡體系中溶液中吸附質的濃度,mg·L-1;Qmax為最大吸附容量,mg·kg-1。盡管可以擴展為多吸附質競爭和多種位點模型,但只采用上述Langmuir模型還不能應對溶液化學組成變化對離子在帶有永久/可變電荷吸附劑表面吸附的影響。將吸附反應過程分為特異性化學反應和非特異性靜電作用是將表面絡合模型機理化的重要一步,分別以位點與吸附質之間的絡合反應模型和靜電模型來表示。離子與顆粒表面的活性官能團可形成內圈配合物(離子直接與吸附劑表面官能團絡合,形成的配合物之間沒有水分子)和/或外圈配合物(離子與吸附劑表面官能團之間包含至少1個水分子)。表面絡合反應的最基礎數學表達式即為Langmuir模型[公式(5)],針對離子在鐵、鋁等金屬(氫)氧化物顆粒表面吸附而發展的模型基本采用了Langmuir模型作為化學絡合模型[26-32]。針對金屬氧化物的表面絡合模型的主要區別在于表面活性位點的類型和數量,以及采用的靜電模型:擴散雙電層模型(Diffuse Double Layer,DDL)、基本斯特恩模型(Basic Stern Model,BSL)和擴展斯特恩模型(Extended Stern Model,ESL)等。在這些模型中,CD-MUSIC(Charge Distribution and Multi-Site Complexation)模型[33-34]是較為先進的離子在氧化物上的表面絡合模型。其位點種類和位點密度源于對礦物表面結構的分析,而離子在氧化物表面的化學形態盡量基于同步輻射技術[35-36]結構和分子動力學預測。光譜技術可以直接揭示離子吸附的微觀機制,包括吸附離子在吸附劑表面形成的化學鍵數目,從而增加了模型的機理性和預測性。除微觀的光譜分析外,宏觀實驗結果(如:電荷滴定曲線、離子強度對吸附的影響、反應過程熱量的變化)亦能揭示離子吸附的機理。以離子強度對吸附的影響為例,離子吸附受體系離子強度變化的影響可以間接反映陽/陰離子在吸附界面形成配合物的類型[37-38]。
土壤有機質是土壤中吸附重金屬離子的重要組分,其化學組成的復雜性和由此帶來的表面位點的多樣性為其表面絡合模型的建立帶來挑戰。計算離子在有機質上的吸附時,常用到的兩種先進的模型是:NICA-Donnan(Non-Ideal Competitive Adsorption)模型[39-40]和 WHAM(Windermere Humic-Aqueous Model)模型[41-42],這兩種模型均考慮了有機質結合位點的異質性。其中WHAM模型[41-42]采用了離散的、非連續分布的處理,即將有機質上的吸附位點分為羧基類和酚羥基類兩大類,每一類位點又分為四種,離子在這四種位點的吸附均以Langmuir模型表示,而同一類的四種位點的KLa值之間保持一定距離分布。NICA模型[39-40]將天然有機質上的羧基類和酚羥基類位點分別視為具有連續分布,其特異性吸附模型:
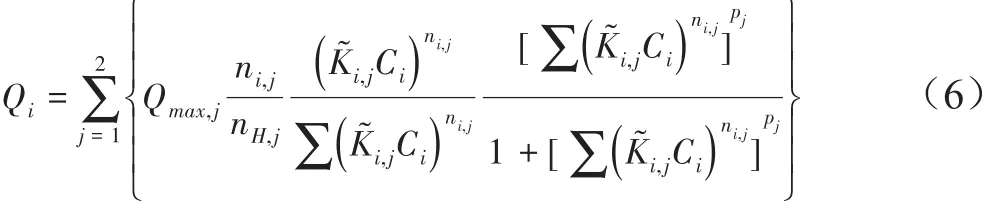
式中:Qi為有機質結合態i離子的含量,mol·kg-1;Qmax,j為有機質顆粒上 j類活性配體的總量,mol·kg-1;ni,j為 i離子結合的j類配體的特異性、非理想性參數(0< ni,j≤1);ni,j/nH,j描述的是離子i化學反應的平均計量系數;K?i,j為平均親和常數;Ci為離子 i在 Donnan相的濃度,mol·L-1;pj為 j類活性配體的化學異質性(0< pj≤1)[40]。NICA模型假設每一類位點的同一離子表面絡合常數(K)是遵循接近正態分布(Sips分布)連續分布。WHAM模型和NICA模型,都結合了Donnan模型作為離子在天然有機質上吸附的靜電模型。不同有機質的Donnan體積大小與腐植酸的性質及體系的離子強度等相關[43-44]。
表面絡合模型(SCMs)在離子形態上預測的發展多經歷從單一介質(金屬氧化物、純化腐植酸等)至天然環境樣品(土壤、水體、沉積物)的過程。目前,針對單一介質的機理性模型的發展已經比較成熟。圖1和圖2分別為Cd在從土壤中純化的胡敏酸(Humic Acid,HA)和富里酸(Fulvic acid,FA)上的吸附[40,45]及Cd、As在針鐵礦上的吸附[46-47],并比較了實驗數據與NICA-Donnan模型和CD-MUSIC模型的計算結果。可以看出,模型可以很好地描述實驗數據。受益于模型的機理性特征,這些模型能夠可靠計算在多種吸附離子存在情況下顆粒表面的相互協同或者競爭作用[48-49]。目前,針對代表天然有機質的HA和FA已經建立了較為完備的NICA-Donnan和WHAM的模型參數。針對常見的重金屬離子在鐵氧化物上的吸附,也有部分CD-MUSIC模型參數可以使用。
2 土壤中重金屬化學形態機理模型的建立
2.1 建立土壤中重金屬化學形態機理模型面臨的挑戰
基于物理化學的基本原理,從分子水平過程出發,建立土壤重金屬化學形態的機理性模型,旨在提升模型的定量性、普適性和預測性。由于土壤體系的復雜性,模型的建立需綜合考慮土壤溶液中的反應、沉淀-溶解和吸附反應等過程,而吸附反應部分的模型建立是土壤重金屬化學形態模型發展的一個關鍵部分。如上所述,在過去研究中很多先進的表面絡合模型的發展使得定量分析離子、小分子物質在膠體顆粒上的吸附得以實現[33,47,50-53]。但是,如何將模型預測由基于單一類型純化的吸附劑的研究體系應用至環境樣品分析面臨諸多挑戰:
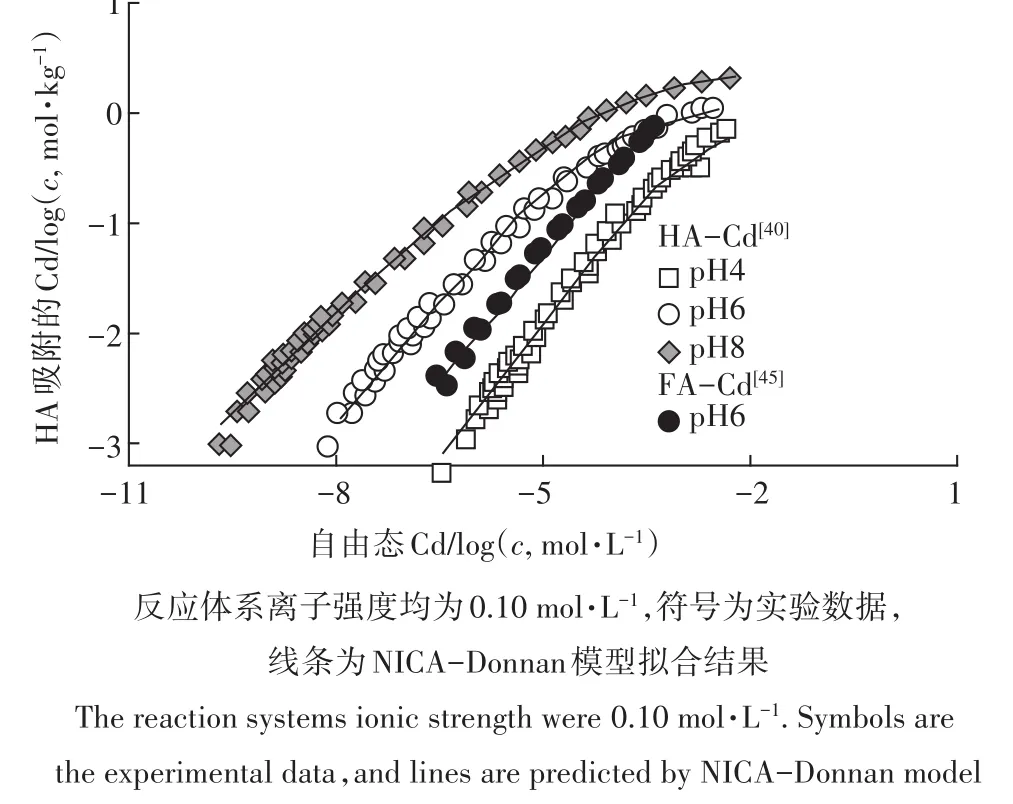
圖1 鎘在HA和FA上的吸附[40,45]Figure 1 Cd binding to HA and FA[40,45]
(1)需要選取某種膠體顆粒作為土壤中一類吸附界面的代表,并確定土壤中該類吸附界面的吸附能力相當于多少選定的模型膠體顆粒。如從土壤中提取的HA和FA常被選作代表土壤中具有重金屬陽離子吸附能力的天然有機質模型膠體顆粒。但是不同土壤的有機質組成、反應活性存在差異[54],同時土壤有機質對重金屬吸附能力也區別于用作模型材料的HA、FA;再如金屬氧化物對重金屬離子的吸附能力不僅取決于其在土壤中的含量,也取決于顆粒的比表面積,即礦物顆粒的粒徑分布。(2)如何取得土壤中重金屬的“活性總量”是建立土壤重金屬化學形態機理模型面臨的另一問題。因為土壤黏土礦物結構和難溶的其他礦物中的重金屬在一般考慮的相關時間內不會參與反應,所以,在以化學平衡為前提的重金屬化學形態模型計算中被視為“非活性部分”需要排除在外。而常用的強酸消解等方法得到的土壤重金屬含量同時包含了“活性”和“非活性”部分的重金屬。(3)面臨的挑戰還包括,離子在代表性膠體顆粒上吸附的研究大多采用單一類型顆粒,而土壤中同時含有多種類型的吸附介質,最直接的模型建立處理方式是假設各類別吸附介質之間沒有交互作用,重金屬在土壤中的吸附總量是在各類界面上吸附量的加和,這種線性疊加模式就是常用的多表面模型(Multi-Surface Model)的基本假設[55]。但是,天然環境樣品中不同類型的顆粒之間可能存在著復雜的交互作用[56],如天然有機質與金屬氧化物顆粒之間的交互作用會影響離子的吸附,導致離子總量不等同于在各類界面上的簡單加和[55]。
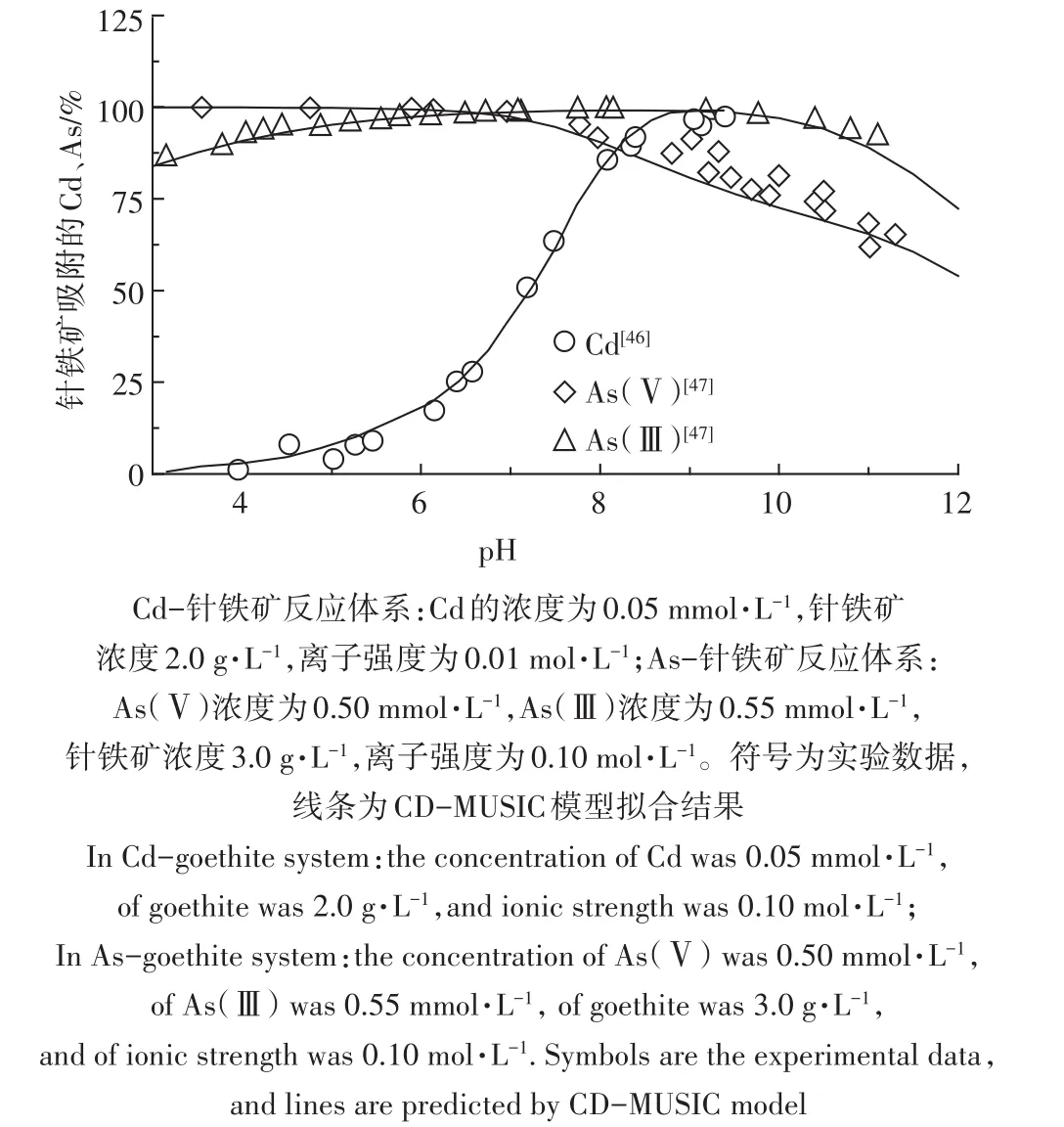
圖2 鎘/砷在針鐵礦上的吸附[46-47]Figure 2 Adsorption of Cd/As(Ⅲ,Ⅴ)onto goethite[46-47]
下面我們對以上問題(1)和問題(2)在已有研究中的處理方式予以回顧,問題(3)將在本文的第三部分結合機理性模型在環境樣品中的應用加以介紹。
2.2 吸附界面含量的確定
2.2.1 土壤有機質
金屬陽離子在有機質上的吸附行為一般由分離純化的HA和FA代表,而通常實驗結果僅包括土壤有機質和可溶性有機質含量,但是有機質的組成、離子結合能力與其來源和土壤時空分布相關[57],所以在模型建立時需要確定土壤有機質離子吸附能力相當于多少HA和FA。常見的一種方法是將一定比例的有機質視為HA或者FA。如Schroder等[58]假定土壤固相體系有機質的50%為HA,土壤溶液中可溶性有機質的40%為FA,其余均為惰性有機質。第二種方法是測定土壤中HA和FA的含量,同時假定在土壤中起作用的有機質為此部分測定的HA或FA[59]。例如:Dijkstra等[60]以快速土壤腐植酸測定方法(Van Zomeren等[61])測得8種沙壤中腐植酸含量占有機質的25%~67%,Groenenberg等[62]測定的沙壤中腐植酸占有機質的81%~87%,Lofts等[63]得到FA和HA占溶解性有機質的63.5%,Ren等[64]研究得出土壤溶液中僅有26.2%的可溶性有機質為腐植酸(主要為FA)。第三種方法是由土壤總陽離子交換量(CEC)減去土壤黏土礦物的CEC得到土壤有機質的電荷密度,并由此折算成HA和FA的含量,如Weng等[65]用這種方法估量了沙壤中有機質的電荷攜帶量相當于含有16%~46%(平均36%)的HA。Gustafsson等[66]通過pH與溶解的Al和Ca間的濃度關系,擬合了14種土壤中腐植酸組分含量,研究結果顯示土壤中活性有機質的含量在17%~84%之間。化學分析及模型擬合測定結果顯示土壤中的活性有機質所占比例在14%~87%之間不等[66-67]。
2.2.2 金屬氧化物
土壤中金屬氧化物多以鐵、鋁(氫)氧化物為主,錳氧化物相對含量較少。因此,大多數計算重金屬化學形態的機理模型只考慮鐵或鐵-鋁的(氫)氧化物對重金屬離子的吸附。但是,有研究指出錳氧化物對金屬離子(如Pb)的吸附也有著很大貢獻[68-69]。Hiemstra等[70]提出離子在鐵、鋁氧化物上的親和常數十分接近,只需考慮不同氧化物比表面積的差異,離子的吸附可以由單一組分氧化物的吸附在模型中代表。鐵、鋁(氫)氧化物在土壤中的總含量一般采取DCB(二亞硫酸鈉-檸檬酸鈉-重碳酸鈉)還原絡合提取方法測定[71],無定形鐵、鋁氧化物含量由草酸銨提取確定[72]。一般將草酸銨提取的鐵、鋁視為無定形氧化物,而DCB與草酸銨提取量之差視為結晶態氧化物[11,73]。結晶態氧化物通常用針鐵礦來代表,無定形氧化物常以水鐵礦來代表[11,70]。Weng等[65]直接假設結晶態的鐵、鋁氧化物的比表面積為100 m2·g-1,而無定形的鐵、鋁氧化物的比表面積為600 m2·g-1。Hiemstra等[70]以磷酸鹽在土壤上的吸附為探針,通過模型反推測了不同土壤中反應活性氧化物的比表面積。
2.2.3 黏土礦物
黏土礦物帶永久性負電荷,對陽離子的吸附以靜電作用為主,同時在其片狀結構的側面也有類似于金屬氧化物表面的活性基團(如鋁醇基)可以吸附陰離子[74]。土壤黏土顆粒(<2 μm)含量可以通過常規土壤性狀分析獲得,并通過礦物組成分析或對樣品當地主要黏土組成的了解,以確定所研究樣品中黏土礦物的主要類型,并由該類黏土礦物的CEC范圍確定土壤黏土顆粒的永久性負電荷密度。Weng等[65]和Rennert等[75]的研究中確定土壤黏土礦物主要成分為伊利石,所以黏土礦物的電荷密度相當于伊利石的陽離子交換量(0.1~0.4 mol·kg-1)。在模擬含氧陰離子在黏土礦物片狀結構側面位點上的吸附時,Gustafsson[76]以水鋁英石(非硅酸鹽類)作為土壤黏土礦物代表,定義了吸附位點特性:鋁醇基的吸附位點為4位點·nm-2。
2.3 重金屬“活性”總量的確定
如上所述,強酸消解能溶解很多一般情況下難以溶解的礦物,所以采用王水消解得到的土壤重金屬含量常會高估“活性”重金屬(如Cd、Pb、Zn、Ni)的含量[77-78]。以2 mol·L-1硝酸提取的Cd和Cu與模型分析相符,但是該濃度硝酸會高估Ni和Zn的土壤吸附量,導致模型計算與測量值之間的差異[55],而以0.4 mol·L-1硝酸提取土壤中的Cd、Cr、Cu、Ni和Zn濃度與模型分析結果能夠很好地吻合[62];也有作者使用螯合劑(如EDTA)提取土壤“活性”重金屬[79-80]。對于有機土(有機質含量大于10%)、森林土和農業耕種土,以EDTA提取與以0.43 mol·L-1硝酸提取的金屬含量結果基本一致[81-82]。由同位素交換反應獲得的“活性”重金屬含量計算得到的溶液中重金屬濃度較利用王水提取的方法相比結果更為準確,尤其是對重金屬Cd、Pb、Zn而言[78]。
土壤中含氧陰離子的“活性”含量常以草酸銨提取測定,其基本原理是在提取過程中隨著無定形金屬氧化物的溶解,吸附在金屬氧化物上的含氧陰離子也釋放出來,如Cui等[56]和Gustafsson[83]用草酸銨提取了土壤中的As作為“活性”As的總量。也有研究者采用酸消解/提取測定土壤“活性”含氧陰離子含量作為模型輸入值總量計算離子的形態分布,但是研究發現土壤酸堿性影響模型預測與提取態離子含量擬合的結果:對于堿性土壤,采用王水消解的As模型預測結果與溶解態As測定值吻合;但在pH<6.8的土壤上,溶解態As高于模型預測值[77]。但是Dijkstra等[60]研究表明在中性及酸性條件下,使用0.43 mol·L-1硝酸提取的As能夠較好地預測可溶態As,而在pH>8時,模型低估了可溶態As。對于硒、銻、鉬酸鹽,使用0.43 mol·L-1硝酸提取能夠很好地預測溶解態硒,但所測溶解態銻與模型預測相比偏高,而所測溶解態鉬偏低[60,62]。因此在模型輸入值確定方面,準確選擇合適的浸提劑定量分析樣品中金屬陽離子、含氧陰離子的濃度尤為重要。
3 機理性模型在土壤重金屬化學形態計算中的應用
近二三十年,機理性化學形態模型在土壤等環境樣品中的應用取得了很大的進展。其中針對金屬陽離子機理性模型的發展領先于含氧陰離子。我們分別以Cd和As作為金屬陽離子及含氧陰離子的代表,舉例說明機理性模型在天然環境樣品重金屬化學形態研究中的應用。
3.1 陽離子:以Cd為例
在有氧條件下,重金屬陽離子在土壤中的分配一般由吸附反應控制。土壤有機質、黏土礦物和金屬氧化物均有一定的陽離子吸附能力。目前常用的方法是基于線性疊加假設的多表面模型(Multi-Surface Model)[65]。多表面模型選取純化腐植酸、合成針鐵礦等模型材料分別代表土壤中的一類吸附界面,根據這些模型材料信息建立的先進的表面絡合模型及模型參數,計算離子在每種界面上的吸附量并予以加和,忽略土壤中各吸附介質的交互作用對離子吸附的影響(多表面模型示意圖如圖3a所示)。一系列的研究表明,多表面模型在很多情況下能夠較好地預測土壤中重金屬陽離子(如Cu2+、Cd2+、Zn2+、Ni2+)的化學形態分布。如:Weng等[65]用NICA-Donnan模型、CD-MUSIC模型、DDL模型和Donnan模型分別計算離子在天然有機質(固相中和可溶性有機質)、晶態和非晶態鐵氧化物,以及黏土硅酸鹽上的吸附,模型預測的Cd2+活度與DMT測定結果比較表明,模型能夠很好地預測Cd2+在土壤中的吸附行為。同樣,Cances等[84]應用相似的模型方法表明,在pH 4~6時模型預測自由態Cd2+與DMT法測定結果基本一致。Dijkstra等[85]在pH 0.4~12的范圍內應用相似的模型方法研究了重金屬在污染土壤中的淋溶特征,結果表明模型基本準確地預測了Cd的淋溶特征。但是,也有研究顯示多表面模型在一定程度上低估了Cd在土壤中的吸附。如:Schroder等[58]研究河灘土壤(pH 5~8)金屬離子的形態分布時,模型預測結果與測量結果相比基本合理,但是模型高估了大約0.5個log單位的溶解態Cd2+(圖4a)。Bonten等[86]在353個非污染和污染土壤上的研究也表明,較高濃度時模型能很好地預測Cd在溶液中的濃度,而在較低濃度時模型平均高估了溶解態Cd2+。類似的報道還有Rennert等[75]的工作(圖4a)。在厭氧環境下,重金屬陽離子可能形成硫化物沉淀,這時模型不但要考慮吸附反應也要考慮硫化物沉淀的形成[87]。
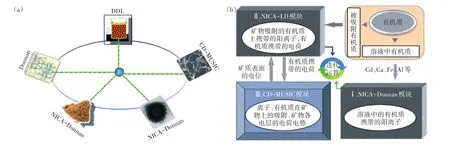
圖3 多表面模型(a)和LCD模型(b)示意圖(b圖修改自Weng等[50])Figure 3 Schematic overview of the multi-surface model(a)and LCD model(b)(b is modified from Weng et al[50])
應用純化膠體顆粒所得到的研究結果顯示,天然有機質與氧化物等礦物表面的交互作用會影響陽離子在有機-礦物復合體系中的吸附[88-90],主要影響機制包括:靜電作用、位點競爭、形成三相復合物(礦物-離子-有機質,礦物-有機質-離子)。目前的研究顯示,基于線性疊加的多表面模型在許多情況下能較為可靠地預測重金屬陽離子(尤其是Cu2+)在土壤中的化學形態分布,這可能是因為:(1)在土壤一般pH范圍內,土壤有機質是陽離子的主要吸附界面,而土壤中與氧化物等礦物直接緊密接觸的有機質只占總有機質的一部分,從而減少了有機質-礦物交互作用對離子吸附的影響;(2)雖然有機質和礦物的交互作用對離子的吸附產生了影響,但是最后的綜合結果與線性疊加的結果接近,比如在FA和針鐵礦的混合體系中,盡管考慮有機質-礦物交互作用的LCD模型計算指出FA和針鐵礦對Cu2+吸附的貢獻受到了兩種吸附劑交互作用的影響,同時形成了針鐵礦-Cu-FA三相復合物,但是Cu2+的吸附總量接近線性疊加結果[90]。但是對于Cd來說,如上所述,一些研究也表明多表面模型可能會低估Cd在土壤中的吸附。除了其他的不確定性以外(如模型輸入值、模型參數的不確定性),天然有機質和礦物的交互作用可能會使得Cd的總吸附量高于線性疊加的結果[91]。
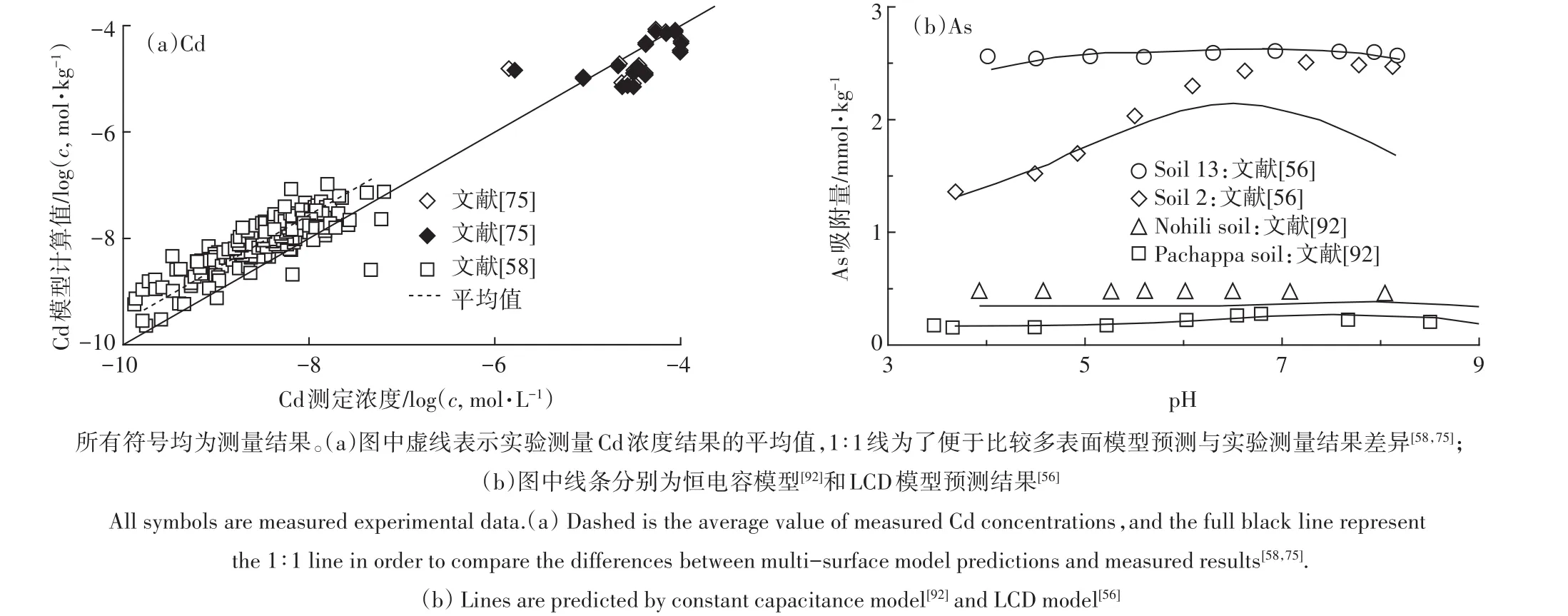
圖4 鎘/砷在土壤上吸附測量結果與模型預測比較Figure 4 The adsorption of Cd and As on soils by comparison with model predictions
3.2 含氧陰離子:以As為例
含氧陰離子的主要吸附界面為金屬氧化物和黏土礦物。陰離子在金屬氧化物上的吸附模型與陽離子在天然有機質上的模型發展基本同步,但是,當應用于天然樣品時,含氧陰離子模型要落后于針對陽離子的模型,其中的一個主要原因就是如何模擬天然有機質對含氧陰離子在礦物表面的競爭吸附作用。一般表面絡合模型只考慮小分子如無機離子在膠體顆粒表面的吸附,但是天然有機質是帶有活性基團的大分子,它在礦物表面的吸附及對含氧陰離子的影響難以在一般表面絡合模型中模擬。Gustafsson[76]應用CD-MUSIC模型研究了As(Ⅴ)在土壤上的吸附,認為土壤中的金屬氧化物(水鐵礦、三水鋁石)和黏土礦物(水鋁英石)是起主要作用的吸附界面,然而在這一模型計算中有機質在礦物上的吸附對As(Ⅴ)吸附的影響被忽略了。事實上,土壤中普遍存在的有機質對陰離子在金屬氧化物等礦物表面的吸附存在著強烈的競爭作用[53,93]。在 Gustafsson[83]另一 CD-MUSIC 研究中,將有機質對As(Ⅴ)在土壤上吸附的影響簡化為礦物表面基團與羧基的絡合反應。與此類似,Hiemstra等[93]應用NOM-CD模型研究了腐植酸對磷在針鐵礦上吸附的影響,與CD-MUSIC模型相比,Hiemstra等[93]在NOM-CD模型計算中加入了以兩個羧基代表的吸附在針鐵礦表面的天然有機質,并允許該羧基發生質子化反應。以羧基代表吸附在礦物表面的有機質可以將有機質對陰離子吸附的影響納入一般表面絡合模型的計算模式中,但是這一處理可能過于簡化,一旦體系pH、Ca、等條件發生變化,模型就不能準確預測吸附的有機質攜帶電荷的變化及對含氧陰離子吸附的影響。
LCD模型結合了NICA模型和CD-MUSIC模型用于計算在天然有機質影響下土壤礦物表面發生的表面絡合反應(LCD模型示意圖如圖3b所示)。Cui等[56]結合LCD模型研究了5種不同土壤As(Ⅴ)吸附與土壤氧化物組成的關系(其中2種土壤實驗結果如圖4b所示),該模型構建方法與Weng等[94]研究磷在土壤上吸附方法相似。具體來說:認為As(Ⅴ)在土壤上的吸附主要是由金屬氧化物主導,以針鐵礦作為土壤吸附含氧陰離子活性表面的典型代表;吸附在土壤金屬氧化物上的天然有機質以分布在第一和第二Stern層的FA來代表,其中,吸附在第一Stern層的FA的羧基(RCOO-)可與針鐵礦表面的單齒配體(≡FeOH)形成內圈配合物(≡FeOOCR),所有的羧基和酚羥基(RO-)可以結合H+、Ca2+、Al3+和Fe3+。吸附的有機質配體(羧基和酚羥基)與表面位點、質子及其他陽離子間的作用以NICA模型進行計算,并假定NICA模型參數和溶液中FA的參數相同。結果表明,在pH 6~8范圍As(V)溶解度的降低主要是由于Ca吸附的促進作用。土壤中磷酸根的吸附強度高于As(Ⅴ),而在針鐵礦上二者的吸附強度類似,這可能是由于土壤氧化物中Al替代Fe降低了對As(Ⅴ)的吸附能力[56]。天然環境中As形態受環境體系氧化還原電位的影響,在還原條件下(如:淹水水稻田),土壤中As(Ⅴ)可還原成As(Ⅲ),同時由于反應速度較慢,As(Ⅴ)和As(Ⅲ)可同時存在。因此在模型預測分析中,需要考慮不同價態的As。Cui等[56]應用HPLCICPMS測定了土壤溶液中As(Ⅴ)與As(Ⅲ)的含量,并用LCD模型計算了As(Ⅴ)和As(Ⅲ)在土壤有機-無機礦物膠體上的吸附。
3.3 計算機軟件
為了增強對復雜環境樣品體系中元素化學形態及遷移的計算能力,人們開發了多種計算機軟件。用于水和土壤等環境樣品中重金屬化學形態計算的常用軟件包括:Visual-MINTEQ[95]、PHREEQC[96]、ECOSAT[97]、WHAM[42]和ORCHESTRA[98]。所有這些軟件的基本原理和構成大體相似,即根據化學反應平衡和質量平衡建立方程式組,由軟件中的方程解鎖核心通過迭代運算求出未知數值。軟件中預定義了不同類型反應的數量關系,如:酸-堿反應、沉淀反應、吸附模型等,使用者可以根據所研究體系的組成特征選擇相關的組分及反應類型,較為方便地去定義具體的化學反應平衡體系[98]。其中,ORCHESTRA軟件采用開放模塊定義某種反應類型的數量關系,用戶在使用過程中可以自由地調整/增加模塊,因而ORCHESTRA軟件增加了使用的靈活性[99]。目前模擬天然有機質-礦物交互作用的LCD模型只在ORCHESTRA環境下運行,而基于線性疊加假設的多表面模型原則上可以在以上所有的軟件環境下進行。至今CD-MUSIC和NICADonnan模型計算均可在ECOSAT、Visual-MINTEQ和ORCHESTRA中完成。
4 總結與展望
針對土壤中重金屬化學形態的定量研究,模型計算可以彌補實驗室分析手段的不足,獲得更為全面和具有預測性的結果。近二三十年,先進的表面絡合模型的發展極大地促進了機理性化學形態模型在環境樣品中的應用。其中,基于線性疊加假設的多表面模型能夠較為成功地預測諸如Cu、Cd、Ni、Zn等重金屬陽離子在環境樣品中尤其是在弱酸性條件下的化學形態分布,但目前多表面模型常低估Pb在土壤中的吸附。含氧陰離子在土壤中的化學形態模型的發展落后于針對陽離子的模型,其主要原因是線性疊加模型不能反映天然有機質對含氧陰離子在礦物表面吸附的競爭作用。近期發展起來的LCD模型是基于非線性疊加假設、涵蓋有機質-礦物交互作用的表面絡合模型,已經被應用到磷、砷、硒這些含氧陰離子在土壤中化學形態分布的研究中。今后的研究可以從以下幾個方面入手:
(1)進一步獲取與完善離子在單一吸附介質上的模型參數,如銻在金屬氧化物上的CD-MUSIC模型參數。
(2)選擇適合的測定“活性”重金屬含量的提取劑,尤其是針對某些類型的土壤比如石灰性土壤、礦區土壤的“活性”重金屬提取劑。
(3)明確適合代表土壤金屬氧化物離子吸附能力的模型材料,如是否可以用實驗室合成的針鐵礦代表土壤中的鐵氧化物。
(4)進一步驗證與發展LCD模型及其在重金屬離子化學形態計算中的應用。
(5)基于LCD模型運行結果,建立更為簡便的有機質-礦物交互作用模型。
(6)進一步擴大重金屬化學形態模型的應用范圍,使其在科學研究以外獲得在污染風險評估、污染環境修復領域更廣泛的應用。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
科技知識動漫(2017年7期)2017-08-09 19:52:45
科技知識動漫(2017年5期)2017-05-11 21:34:16
科技知識動漫(2017年4期)2017-04-15 22:24:55
科技知識動漫(2017年2期)2017-02-06 20:59:46
光學精密工程(2016年6期)2016-11-07 09:07:19
科技知識動漫(2016年10期)2016-10-18 20:35:00
核科學與工程(2015年4期)2015-09-26 11:59:03
