月見草油氣相色譜指紋圖譜研究
2018-08-10 02:17:04王慧竹徐麗華
中國釀造 2018年7期
關(guān)鍵詞:方法
王慧竹,陳 帥*,石 琳,韓 緒,張 龍,張 敏,徐麗華
(1.吉林化工學院 化學與制藥工程學院,吉林 吉林 132022;2.吉林大學 藥學院,吉林 長春 130021)
月見草(Oenothera biennisL.)為柳葉菜科月見草屬1~2年或多年生草本植物,其原產(chǎn)于北美地區(qū),在我國主產(chǎn)于東北三省[1],月見草全身是寶,月見草揮發(fā)油可食用,花可提取芳香油,莖皮纖維可制繩,根是解熱、消炎的良藥,莖和葉有清熱解毒、鎮(zhèn)痛的功效[2-3],種子可榨油食用和藥用,其中月見草油是其主要成分,主要由亞油酸、油酸、棕櫚酸、亞麻酸多種脂肪酸和維生素等組成[4],具有良好的降脂、降糖、減肥、抗炎、抗氧化、抗血栓功能[5-7],對老年慢性疾病具有醫(yī)療保健作用。隨著對月見草油藥理作用的進一步認識,全世界對月見草油的需求也日益增長。目前對月見草油的研究主要集中在提取工藝、成分分析、藥理作用等方面[8-10]。
中藥指紋譜圖技術(shù)是指用色譜、光譜檢測方法獲得中藥成分的譜圖,然后對圖譜進行“過濾”、簡化等加工處理,從而獲得穩(wěn)定和專屬的特征指紋圖譜,以此來控制藥材質(zhì)量[11-13]。
本實驗以月見草油為研究對象,采用氣相色譜法結(jié)合柱前甲酯衍生化法,建立不同產(chǎn)地月見草油的指紋圖譜,通過國家藥典委員會指紋圖譜評價軟件計算相似度,并結(jié)合化學計量學中的聚類分析和主成分分析,探討了不同批次月見草油差異的來源,以期為月見草油的質(zhì)量控制提供科學依據(jù)。
1 材料與方法
1.1 材料與試劑
來源于不同產(chǎn)地的月見草藥材購自吉林各大藥材零售商,經(jīng)吉林化工學院陳帥副教授鑒定為月見草正品,月見草藥材的批次編號、批號及產(chǎn)地信息匯總情況見表1。
石油醚、環(huán)己烷(分析純)、對照品:γ-亞麻酸甲酯(批號:20110723,純度≥98%)、亞油酸甲酯(批號:20176307,純度≥98%)均由武漢華士特工業(yè)生物技術(shù)開發(fā)有限公司提供。
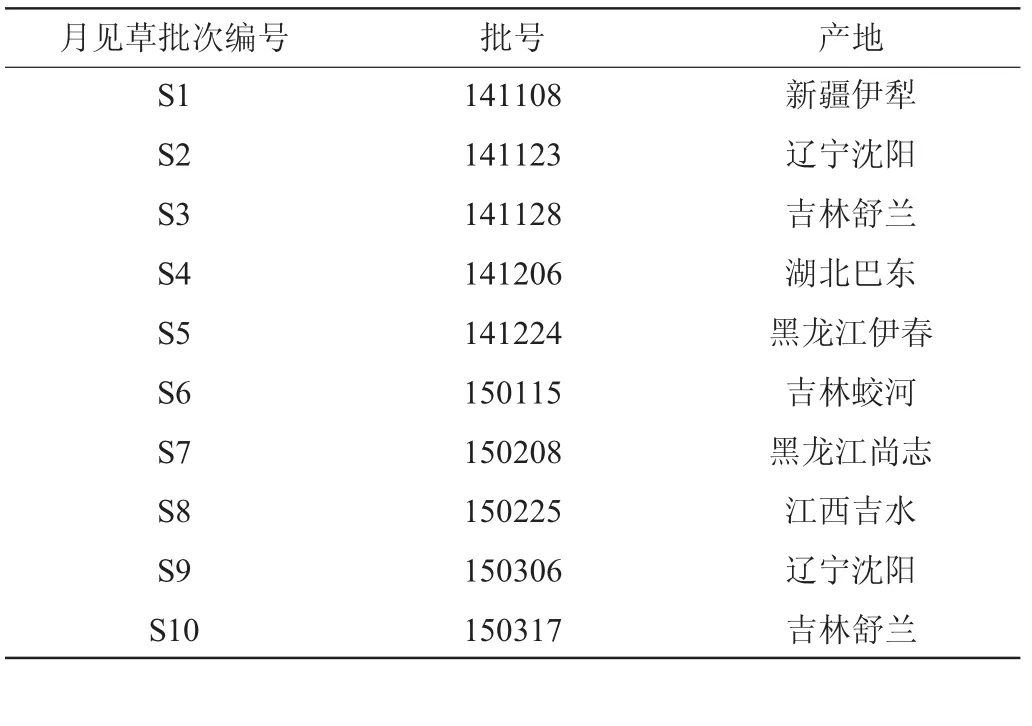
表1 月見草來源及編號Table 1 Sources and number ofOenothera biennis
1.2 儀器與設備
FL 9720氣相色譜儀:浙江福立分析儀器有限公司;AUY220型電子分析天平:日本島津公司;HP-5毛細管氣相色譜柱:美國安捷倫科技有限公司;氫火焰離子化檢測器:浙江福立分析儀器有限公司;SHC型高純氫氣發(fā)生器、JK-5L型純凈空氣泵:山東賽克賽斯氫能源有限公司;BJ-100植物粉碎機:上海拜杰實業(yè)有限公司;WSP-100恒溫水浴鍋:上海申生科技有限公司;SHZ-DⅢ循環(huán)水真空泵:鞏義市予華儀器有限責任公司;SHZ-A水浴恒溫振蕩器:常州國旺儀器有限公司;H1850臺式高速離心機:湖南長沙湘儀離心機廠。
1.3 方法
1.3.1 色譜條件的優(yōu)化方法
GC9720色譜柱(0.25 mm×30 m×0.25 μm);分流比30∶1;檢測器溫度280℃;載氣高純氮氣;柱流量1.5 mL/min;氫氣流量30mL/min;空氣流量300 mL/min;進樣量2 μL;柱溫箱溫度:在實驗條件優(yōu)化過程中共篩選了3個升溫程序。
A:初始溫度50℃,保持2min,以10℃/min升溫至100℃,保持2 min,以15℃/min升溫至140℃,保持5 min,以8℃/min升溫至200℃,保持10 min。
B:初始溫度55℃,保持2min,以7℃/min升溫至120℃,保持3 min,以16℃/min升溫至180℃,保持5 min,以8℃/min升溫至220℃,保持12 min。
C:初始溫度55℃,保持3min,以5℃/min升溫至100℃,保持2 min,以20℃/min升溫至160℃,保持3 min,以3℃/min升溫至230℃,保持15 min。1.3.2供試品溶液制備方法
取自然晾干的月見草適量,粉碎,過50目篩,準確稱量100 g,置于2 000 mL圓底燒瓶中,加正己烷1 200 mL,水浴加熱回流提取2次,每次1.5 h,合并提取液,減壓回收溶劑,得月見草油。由于正己烷直接萃取的月見草提取液中可能會混有部分長鏈脂肪酸或不飽和脂肪酸,若直接進氣相分析,會污染毛細柱,因此需將在進樣前對其進行衍生化,本文采用氫氧化鉀-甲醇溶液進行脂肪酸甲酯化的方法進行樣品處理,在處理過程中,分別對氫氧化鉀-甲醇溶液濃度、加入量、反應時間3個因素進行考察。具體操作為:取月見草油約50 mg,置于具塞離心試管中,加入5 mL正己烷中,然后分別加入濃度為(1 mol/L、2 mol/L、3 mol/L)的KOH甲醇溶液[14](0.5 mL、1.0 mL、2.0 mL),室溫振蕩反應(2 min、3 min、5 min、10 min),以1 200 r/min的轉(zhuǎn)速進行離心,取上層有機相,置于10 mL容量瓶中,正己烷定容,搖勻,進樣前經(jīng)0.45 μm微孔濾膜過濾。
1.3.3 標準品溶液的制備
分別精密稱定γ-亞麻酸甲酯0.003 0 g、亞油酸甲酯0.005 0 g,分別置于兩個5 mL棕色容量瓶中,加正己烷溶解并定容至刻度,即得γ-亞麻酸甲酯、亞油酸甲酯標準品貯備液;分別精密移取上述兩個標準品貯備液0.4 mL,分別置于同一個5 mL容量瓶中,即得體積分數(shù)分別為48 μL/mL和80 μL/mL的γ-亞麻酸和亞油酸混合標準品溶液,冷藏備用。
1.3.4 數(shù)據(jù)處理
指紋圖譜相似度計算采用國家藥典委員會“中藥色譜指紋圖譜相似度評價系統(tǒng)軟件”2012.130723版本,計算方法采用均值法;聚類分析采用SPSS20.0軟件,以月見草油指紋圖譜各共有峰面積為數(shù)據(jù)源,采用系統(tǒng)聚類中的組間連接方法,樣品距離度量采用平方Euclidean距離進行計算;利用SIMCA 14.1軟件,以不同批次月見草油各共有峰的峰面積為自變量,進行主成分分析。
2 結(jié)果與分析
2.1 條件優(yōu)化
在柱溫箱溫度優(yōu)化過程中因C升溫程序的色譜圖中,各色譜峰分離度良好,色譜峰分布均勻,故最終選擇C升溫程序進行實驗;供試品溶液甲酯化試驗結(jié)果表明,在相同的色譜條件下,加入2 mol/L的KOH甲醇溶液1.0 mL,室溫振蕩反應5 min,色譜峰較多、峰面積較大,且樣品處理時間合適,因此在供試品溶液制備過程中,采用向樣品溶液中加入2 mol/L的KOH甲醇溶液1.0 mL,室溫振蕩反應5 min的方法進行甲酯化。將表1中各批月見草藥材分別按“1.3.2”項下優(yōu)化出的最佳方法制備供試品溶液,并按“1.3.1”項優(yōu)化出的最佳色譜條件測定,記錄結(jié)果見圖1(圖中顯示樣品S4的色譜圖)。
2.2 月見草油指紋圖譜的建立
2.2.1 參比峰的選擇
通過氣相色譜法對10批月見草油進行測定,在樣品的氣相色譜圖(圖1)中,亞油酸甲酯(4號峰)色譜峰的峰面積較大,與相鄰色譜峰的分離度較好,且為10批月見草油指紋圖譜所共有,因此,本研究將其設為參照峰,用以計算其他共有峰的相對保留時間和相對峰面積。
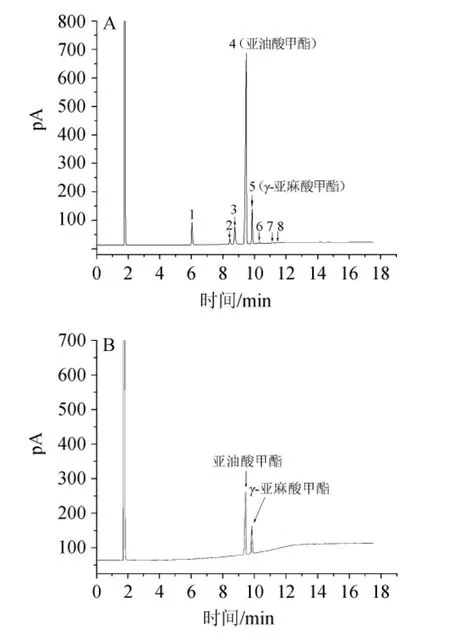
圖1 月見草油樣品S4(A)和混合標準品(B)氣相色譜圖Fig.1 GC chromatogram ofOenothera biennisoil sample S4(A)and mix standards(B)
2.2.2 指紋圖譜的建立及相似度評價
將所得的10批月見草揮發(fā)油GC圖譜以AIA格式依次導入中國藥典委員會研制的中藥色譜指紋圖譜相似度評價系統(tǒng)(2012.130723版),建立了月見草揮發(fā)油GC指紋圖譜疊加模式色譜圖,結(jié)果如圖2所示。將10批月見草油按平均值法生成對照指紋圖譜,通過對各批次的指紋圖譜的相似度計算,結(jié)果見表2。
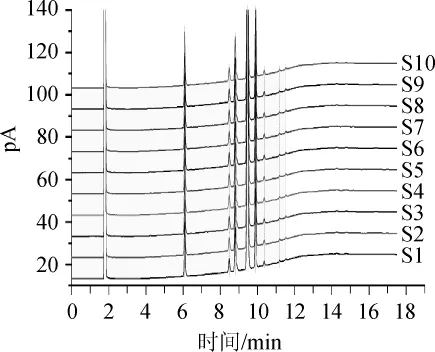
圖2 10批月見草油GC指紋圖譜疊加圖Fig.2 GC fingerprint superposition of 10 batches of Oenothera biennisoil
由圖2可知,以樣品S4藥材圖譜作為參照譜進行指紋匹配,確定了8個共有峰。由表2可知,以10批月見草油按平均值法生成對照指紋圖譜相似度為1進行計算,10批藥材的相似度在0.914~0.989范圍內(nèi)。
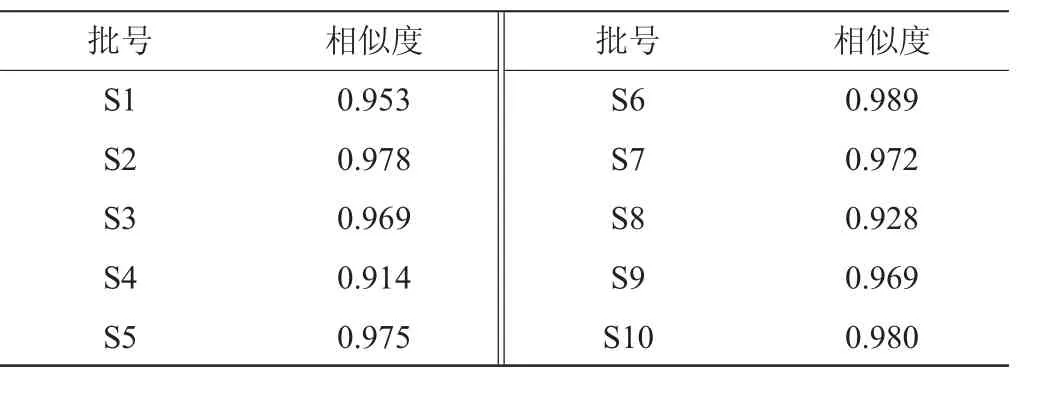
表2 10批月見草揮發(fā)油GC指紋圖譜相似度Table 2 Similarity of GC fingerprint of the 10 batches of Oenothera biennisoil
2.3 方法學考察
2.3.1 精密度試驗
取同一供試品溶液(樣品S4),按“1.3.1”項下方法優(yōu)化出的最佳色譜條件,連續(xù)進樣5次,記錄色譜圖,以4號峰(亞油酸甲酯)為參照峰,計算各共有指紋峰的相對保留時間和相對峰面積結(jié)果相對標準偏差(relative standard deviation,RSD)分別見表3和表4。
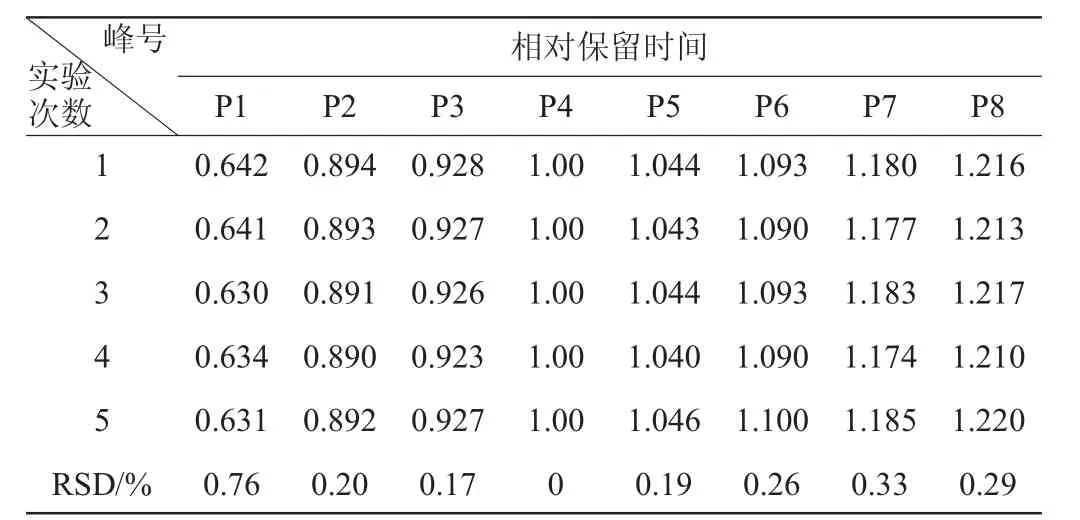
表3 相對保留時間的精密度試驗結(jié)果Table 3 Experimental results of precision for relative retention time
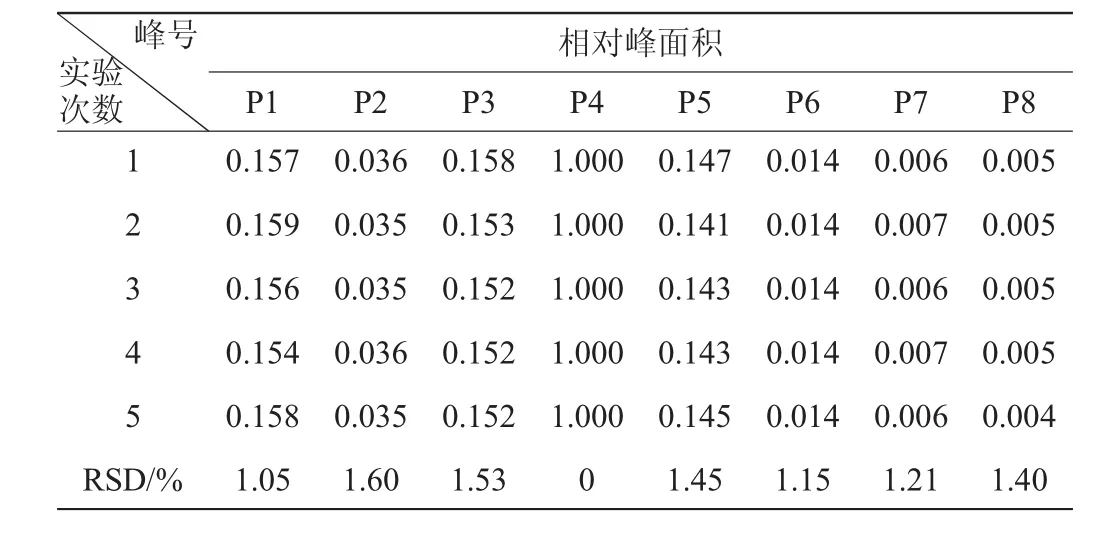
表4 相對峰面積的精密度試驗結(jié)果Table 4 Experimental results of precision for relative peak area
由表3可知,各共有峰相對保留時間的相對標準偏差RSD<0.76%;由表4可知,相對峰面積的RSD<1.60%,表明方法的精密度良好。
2.3.2 穩(wěn)定性試驗
取同一供試品(樣品S5)溶液,按“1.3.1”項下方法優(yōu)化出的最佳色譜條件,分別于2 h、4 h、6 h、8 h、12 h進樣,記錄色譜圖,以4號峰(亞油酸甲酯)為參照峰,計算各共有指紋峰的相對保留時間和相對峰面積見表5和表6。
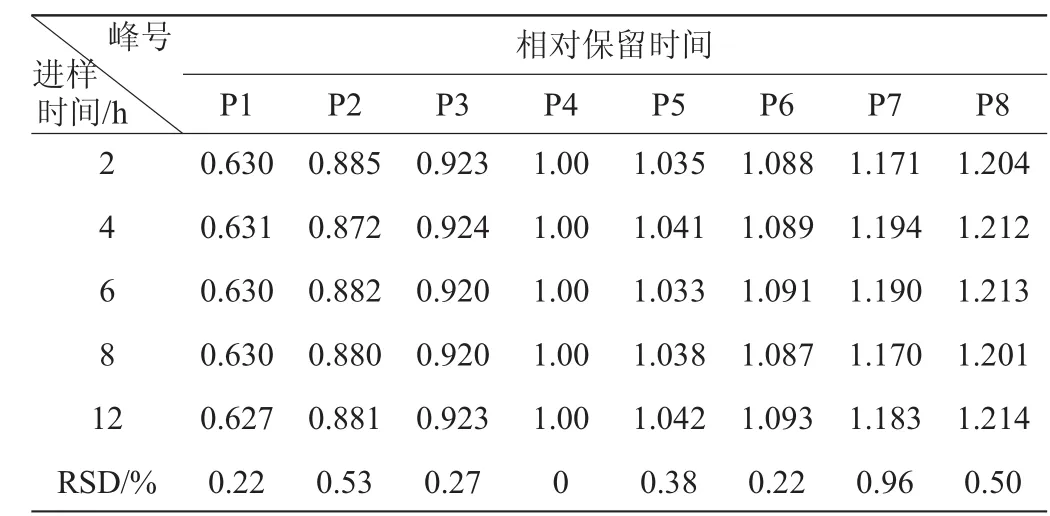
表5 相對保留時間的穩(wěn)定性試驗結(jié)果Table 5 Experimental results of stability for relative retention time
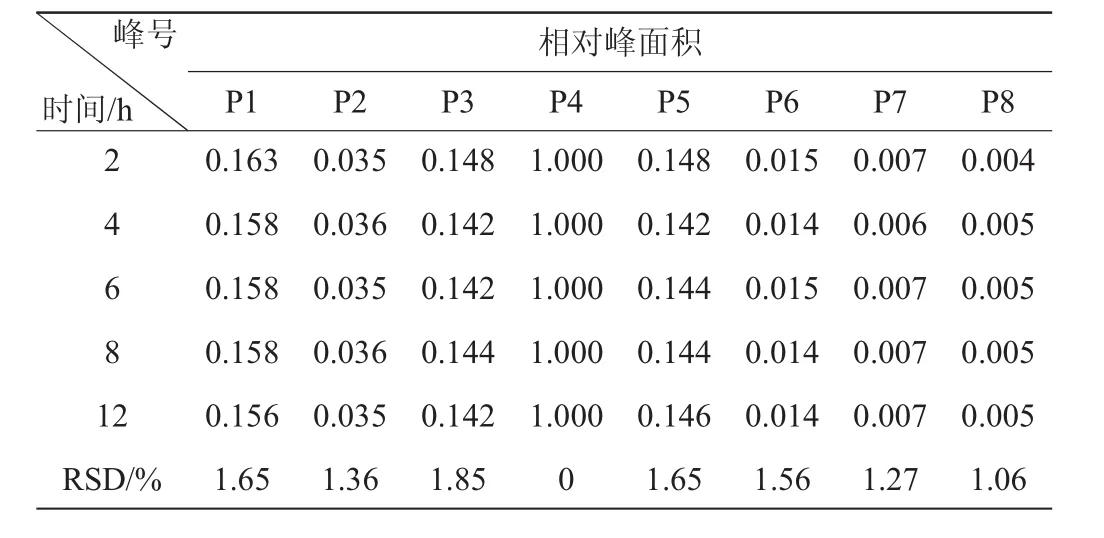
表6 相對峰面積的穩(wěn)定性試驗結(jié)果Table 6 Experimental results of stability for relative peak area
由表5、表6可知,各共有峰相對保留時間RSD<0.96%,各共有峰相對峰面積RSD<1.85%。表明供試品溶液在12 h內(nèi)穩(wěn)定。
2.3.3 重復性試驗
取同一批(樣品S4)月見草藥材5份,按“1.3.2”項下方法優(yōu)化出的最佳供試品溶液制備方法平行制備5份月見草油供試品溶液,按“1.3.1”項下方法優(yōu)化出的最佳色譜條件,依次測定,記錄色譜圖,以4號峰(亞油酸甲酯)為參照峰,計算各共有指紋峰的相對保留時間和相對峰面積見表7和表8。
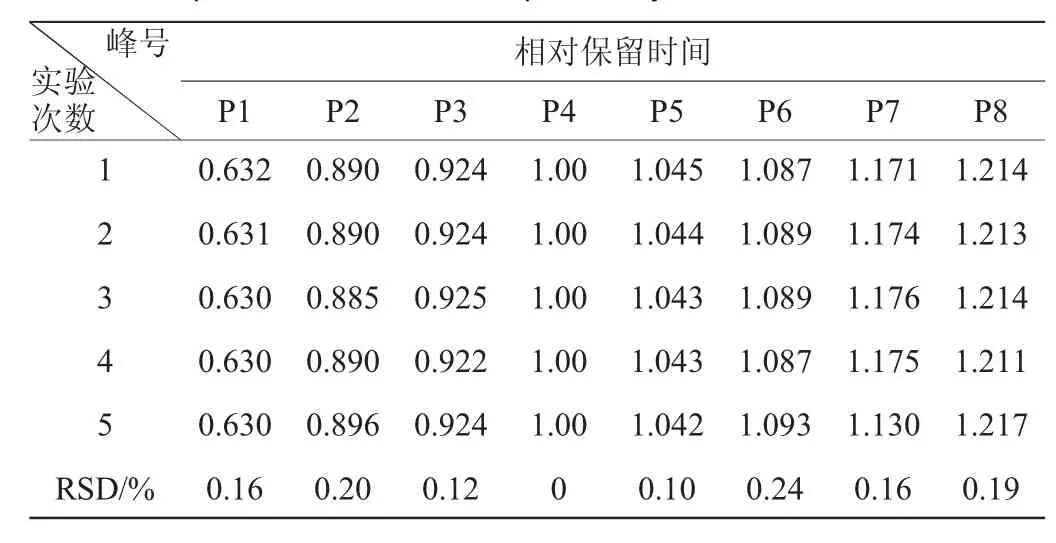
表7 相對保留時間的重復性試驗結(jié)果Table 7 Experimental results of repeatability for relative retention time
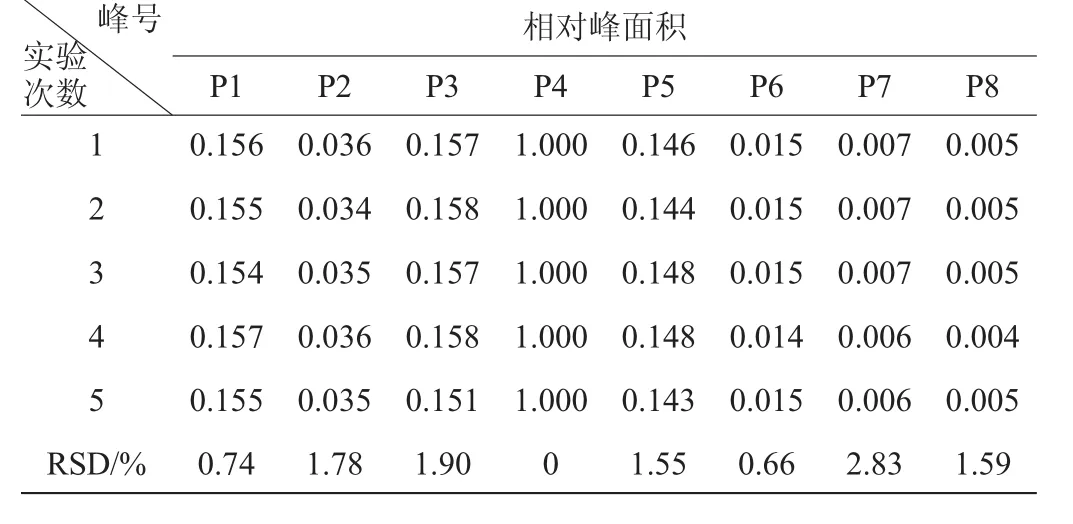
表8 相對峰面積的重復性試驗結(jié)果Table 8 Experimental results of repeatability for relative peak area
由表7、表8可知,各共有峰相對保留時間RSD<0.24%,各共有峰相對峰面積RSD<2.83%,表明該方法具有較好的重復性。
2.4 月見草油指紋圖譜的聚類分析
本研究以月見草油指紋圖譜各共有峰面積為數(shù)據(jù)源[15],對10批月見草油的GC指紋圖譜進行聚類分析,采用系統(tǒng)聚類中的組間連接方法,樣品距離度量采用平方Euclidean距離,得到10批月見草油指紋圖譜的聚類分析樹狀圖,結(jié)果如圖3所示。
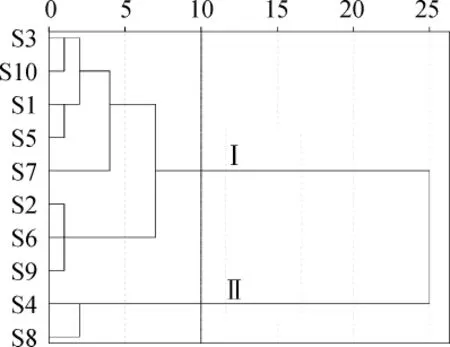
圖3 10批月見草油指紋圖譜的聚類分析樹狀圖Fig.3 Cluster analysis tree for 10 batches ofOenothera biennisoil
聚類分析圖反映的是各樣品的相似程度,樣品間的分類距離值越小則樣品間的差異越小,反之則越大。由圖3可知,當分類距離取10時,10批月見草樣品被分為2類,即Ⅰ和Ⅱ類,其中S4和S8為II類,其余樣品為I類。
2.5 主成分分析
以主成分特征值>1作為選擇主成分的依據(jù),獲得2個主成分,主成分分析結(jié)果如圖4所示。
由圖4可知,累計均方差為96.5%,表明引入的2個主成分可以表達96.5%的原變量信息,從圖4A中可以看出,10批樣本大致分為兩組,分別標記為I和II。其中,S4和S8為II組,其余樣品為I組,由于S4和S8樣品來源分別為湖北巴東和江西吉水,而其余樣品均來自北方寒冷地區(qū),結(jié)合分析結(jié)果,地理位置與氣候條件的差異對月見草質(zhì)量存在一定程度的影響。從圖4B中可以看出1,2,3,4,5,6號峰對PC1有較大的貢獻,而7,8號峰對PC2有較大的貢獻。
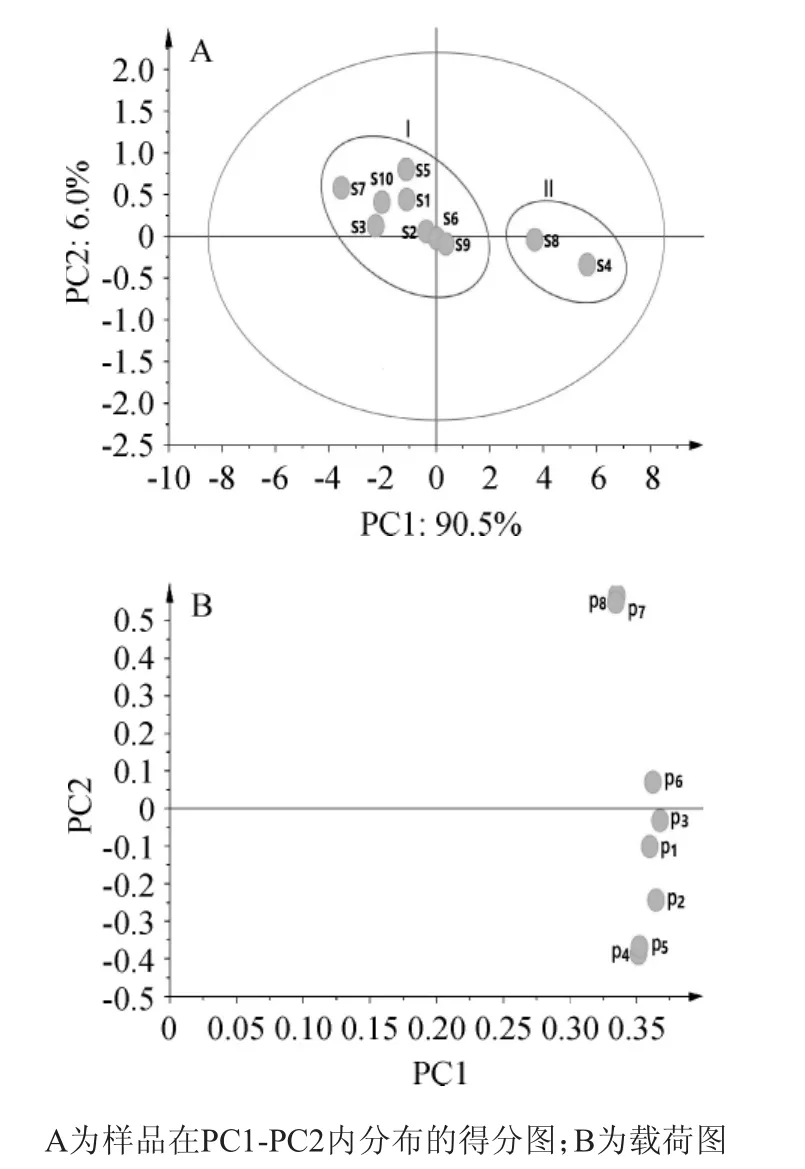
圖4 不同批次月見草油指紋圖譜的主成分分析結(jié)果Fig.4 Results of principle components analysis of fingerprint of oil fromOenothera bienniswith different batches
3 結(jié)論
在最優(yōu)的樣品處理方法和色譜條件下,建立了月見草油的指紋圖譜,經(jīng)方法學考察,該方法精密度、重復性和穩(wěn)定性良好,均符合指紋圖譜規(guī)定要求。通過對指紋圖譜分析,確證了8個共有峰,10批月見草油的相似度為0.914~0.989。考慮到中藥這一復雜體系中的不同的化學物質(zhì)之間可能存在著一定的關(guān)聯(lián)性,因此聚類分析和主成分分析被應用于此研究中,并且聚類分析和主成分分析的結(jié)果一致,所選樣本在兩種方法下均被分成了兩類,同時主成分分析結(jié)果表明1、2、3、4、5、6號峰為影響月見草油指紋圖譜相似度評價的主要因素(累計方差為90.5%)。由于受實驗條件所限,本實驗只鑒定出4號峰和5號峰分別為亞油酸甲酯和γ-亞麻酸甲酯,其它色譜峰代表的化學成分需進一步研究。本研究建立的月見草油指紋圖譜結(jié)合化學計量學的方法,為月見草油質(zhì)量控制提供了依據(jù)。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
