我國黃淮麥區10個短體線蟲樣品種類的分子鑒定
2018-08-17 02:10:06劉海璐王暄李紅梅李艷霞薛博文馬居奎
中國農業科學 2018年15期
劉海璐,王暄,李紅梅,李艷霞,薛博文,馬居奎
?
我國黃淮麥區10個短體線蟲樣品種類的分子鑒定
劉海璐,王暄,李紅梅,李艷霞,薛博文,馬居奎
(南京農業大學植物保護學院/農作物生物災害綜合治理教育部重點實驗室,南京 210095)
【目的】短體線蟲(spp.)是植物根系內遷移性寄生線蟲,可引起許多作物的根腐線蟲病,給世界農業生產造成了極大的危害。為明確我國黃淮麥區與禾谷孢囊線蟲復合侵染小麥的短體線蟲種類,本研究對采自黃淮4省麥區的10個短體線蟲樣品進行種類的分子鑒定,分析種群系統進化關系及種內遺傳變異,以期為我國小麥根部線蟲病害的綜合防治提供指導。【方法】對采自江蘇、安徽、河南和山東4省小麥孢囊線蟲發病田中的10個小麥短體線蟲樣品進行線蟲分離,從各樣品中隨機挑取5條短體線蟲,分別提取單條線蟲DNA,擴增rDNA 18S片段并進行測序比對,選取序列有代表性的2個DNA樣本進一步擴增其rDNA 28S D2-D3區以及mtDNA-COI基因片段,經序列比對分析后,利用MEGA4.0軟件采用鄰接法分別構建基于rDNA 18S、28S D2-D3和mtDNA-COI序列的系統進化樹,通過聚類關系及相似度分析確定線蟲種類,同時利用種特異性引物進行驗證。【結果】擴增挑取的50條短體線蟲的rDNA 18S區片段,測序得到片段長度在857—935 bp,BLAST比對分析揭示部分樣品可能為短體線蟲的混合種群;進一步測定的20個代表性DNA樣本的rDNA 28S D2-D3區和mtDNA-COI基因的片段長度分別在771—784 bp和415—417 bp;系統進化樹以及相似度分析揭示我國黃淮流域4省麥區10個短體線蟲樣品中有咖啡短體線蟲(e)、落選短體線蟲()和斯克里布納短體線蟲(),其中,江蘇沛縣樣品JS2和山東濰坊樣品SD1是落選短體線蟲單一侵染樣品,河南永城樣品HN2和安徽淮北樣品AH3是咖啡短體線蟲單一侵染樣品,安徽蕭縣樣品AH2、AH5和淮北樣品AH4以及河南永城樣品HN1和HN3均為落選短體線蟲和咖啡短體線蟲的混合侵染樣品,江蘇徐州樣品JS1是落選短體線蟲和斯克里布納短體線蟲的混合侵染樣品。用SCAR特異性引物擴增20個短體線蟲單條DNA樣本,結果顯示,用落選短體線蟲特異性引物PNEG-F1/D3B5能夠從JS1-2、JS2-1、JS2-2、AH2-2、AH4-1、AH5-1、HN1-2、HN3-2、SD1-1和SD1-2等10個樣本中擴增出140 bp的單一條帶,用咖啡短體線蟲引物PC1/PC2能夠從AH2-1、AH3-1、AH3-2、AH4-2、AH5-2、HN1-1、HN2-1、HN2-2和HN3-1等9個樣本中擴增出630 bp的單一條帶,用斯克里布納短體線蟲引物PsF7/PsR7從JS1-1中擴增出130 bp的單一條帶,種類鑒定結果與上述序列分析結果相一致。【結論】我國黃淮流域4省小麥孢囊線蟲發病田中的短體線蟲種類有咖啡短體線蟲、落選短體線蟲和斯克里布納短體線蟲,其中落選短體線蟲是優勢種,證實了短體線蟲不同種群復合侵染小麥的現象較為普遍。基于mtDNA-COI基因構建的系統進化樹可以有效區分短體線蟲的近緣種,相比rDNA 18S和28S基因更適于作為短體線蟲種類鑒定的分子靶標。
短體線蟲;種類鑒定;rDNA;mtDNA;系統進化;SCAR
0 引言
【研究意義】短體線蟲(Pratylenchusspp.)是一類分布廣泛、寄主眾多的遷移性植物內寄生線蟲,可侵染植物根部組織,導致根系表皮破裂、塊莖內部腐爛等;此外,短體線蟲造成的根部傷口可為植物病原真菌和細菌侵入提供有利條件,誘發復合侵染,引起作物減產和農產品品質下降[1-2]。目前短體線蟲屬有效種已達101個[3-4],我國已報道的短體線蟲有20多種,危害的作物種類包括小麥、玉米、大豆、花生、棉花、苧麻、馬鈴薯、山藥、草莓、石榴、煙草等[5]。筆者實驗室在調查我國黃淮麥區孢囊線蟲發生分布過程中[6-7],發現短體線蟲與孢囊線蟲復合侵染小麥根系的現象普遍存在,這種復合侵染對于抗線蟲品種的布局具有極大的影響,因此,明確河南、安徽、山東和江蘇4省小麥短體線蟲的種類,對于指導我國小麥孢囊線蟲病和根腐線蟲病一體化綜合治理具有重要意義。【前人研究進展】國外已有大量關于短體線蟲危害麥類作物的報道,種類主要包括六裂短體線蟲()、盧斯短體線蟲()、落選短體線蟲()、穿刺短體線蟲()、斯克里布納短體線蟲()、桑尼短體線蟲()和玉米短體線蟲()等,其中對小麥危害較大是落選短體線蟲、穿刺短體線蟲和桑尼短體線蟲[8-9]。我國安徽、四川、廣西、河南和西藏等地也有短體線蟲危害小麥的報道,已知種類包括落選短體線蟲、穿刺短體線蟲、咖啡短體線蟲(e)、玉米短體線蟲、盧斯短體線蟲、敏捷短體線蟲()和草地短體線蟲()等[10-15]。近年筆者實驗室在黃淮麥區開展的田間調查中,發現同一田塊存在多種短體線蟲混合發生的現象比較普遍,傳統的線蟲形態學鑒定需要有足夠數量的成蟲用于測計和形態學特征比較,尤其當一個樣品中同時存在多種短體線蟲時,首先需要將不同的種區分開再依據形態特征進行鑒定,因此操作較為繁瑣且鑒定周期相對較長。而分子生物學技術的應用很好的解決了上述難題,Pinochet等[16]最早利用隨機擴增多態性DNA(random amplified polymorphic DNA,RAPD)技術鑒定傷殘短體線蟲(),Ouri等[17]利用限制性片段長度多態性(restriction fragment length polymorphism,RFLP)技術將短體線蟲7個種區分開,此后,特征序列擴增區(sequence-characterized amplified regions,SCAR)[18-21]、環介導等溫擴增(loop-mediated isothermal amplification,LAMP)[22]、DNA條形碼(DNA barcoding)[23]等分子技術也相繼應用于短體線蟲的種類鑒定。其中,SCAR技術應用最為廣泛,目前包括咖啡短體線蟲、落選短體線蟲、斯克里布納短體線蟲、盧斯短體線蟲、桑尼短體線蟲、玉米短體線蟲、傷殘短體線蟲等種類均有特異性引物報道[18-21]。此外,基于核糖體DNA(rDNA)和線粒體DNA(mtDNA)保守區序列的分析,同樣對短體線蟲種類鑒定、系統進化及遺傳變異分析具有重要意義,Subbotin等[24]比較了短體線蟲的rDNA 18S和28S D2-D3區序列,證實了短體線蟲的28S D2-D3區比18S具有更高的種間變異度,28S D2-D3區基因片段更適合作為分子鑒定的靶標;王金成等[25]比較分析了短體線蟲rDNA的ITS區和28S D2-D3區序列,同樣認為28S D2-D3區靶標更適用于短體線蟲種類的檢測;而Janssen等[4]通過分析rDNA 28S、ITS以及mtDNA-COI基因序列澄清了穿刺短體線蟲、偽短體線蟲()和鈴蘭短體線蟲()分類地位的長期爭議。當前GenBank數據庫中已經積累了大量的短體線蟲rDNA 18S、28S D2-D3、ITS以及mtDNA-COI 序列信息,為利用上述基因進行序列比對分析、系統發育樹構建、種內遺傳變異分析等提供了極大的便利。【本研究切入點】由于短體線蟲近緣種間的形態特征非常相似,且種內形態變異較大[1],傳統的形態學鑒定方法費時費力,對混合群體的鑒定可能會存在誤差,而分子鑒定方法操作相對簡單、快捷。目前國內有關小麥短體線蟲種類分子鑒定的系統研究較少。本研究對采自黃淮流域河南、安徽、山東和江蘇4省小麥孢囊線蟲發病田的10個小麥短體線蟲樣品進行線蟲分離,提取單條線蟲DNA,分別利用rDNA和mtDNA序列比對分析、基于SCAR-PCR的種特異性引物擴增兩種方法鑒定短體線蟲的種類。【擬解決的關鍵問題】利用單條線蟲DNA擴增rDNA 18S、28S D2-D3以及mtDNA-COI片段,對擴增片段進行測序和比對分析、系統進化樹構建以及序列相似度分析,明確我國黃淮麥區短體線蟲的種類發生情況,為今后我國小麥根部線蟲病的防治提供理論指導。
1 材料與方法
1.1 樣品采集與線蟲分離
于2016年12月冬小麥分蘗期和2017年4—5月抽穗楊花期,對我國黃淮流域江蘇、安徽、河南、山東4省小麥孢囊線蟲發病田的小麥根腐線蟲病發生情況進行調查,采集小麥根系及根際土壤樣品共10份,具體地理信息見表1。采用淺盤法分離樣品中的線蟲,收集線蟲懸浮液,在體視顯微鏡下觀察線蟲的形態特征。根據短體線蟲的形態特征并結合文獻資料核對,對樣品中的短體線蟲進行初步的鑒定。從各樣品中隨機挑取5條短體線蟲,用于DNA分子鑒定。
1.2 單條線蟲的DNA提取與分子片段的擴增測序
單條線蟲的DNA提取參考宋志強等[26]的方法。將線蟲挑入滅菌ddH2O水滴中清洗1—2次后,挑取單條線蟲放入加有16 μL ddH2O和2 μL 10×PCR Buffer(Mg2+free)的200 μL PCR管中,液氮中冷凍2 min后,65℃處理1 min,重復3次;加入2 μL 10 μg·μL-1蛋白酶K,65℃溫育1 h,95℃處理10 min,在-20℃條件下保存備用。
1.3 rDNA與mtDNA通用引物擴增測序
以短體線蟲單條DNA樣本為模板,分別采用rDNA 18S、28S D2-D3區以及mtDNA-COI等片段通用引物[27-29]進行擴增(表2),PCR反應體系:DNA模板1 μL,5 μmol·L-1的上游引物和下游引物各2 μL,2×Ex Taq Mix 12.5 μL,ddH2O補足至25 μL。擴增條件:94℃預變性4 min;94℃變性30 s,52—55℃(按引物不同)退火30 s,72℃延伸1 min,共40個循環;72℃延伸10 min。PCR產物在1.2%瓊脂糖凝膠上電泳檢測,擴增產物切膠回收后連接至PMD19-T載體,轉化大腸桿菌DH5,經菌落PCR驗證后送至南京擎科生物公司測序。

表1 黃淮麥區10個短體線蟲樣品的采集信息和種類鑒定

表2 本研究所用引物信息
1.4 序列比對分析和系統進化樹構建
使用ContigExpress軟件將獲得的序列進行拼接,用BLAST進行序列比對分析后,提交GenBank獲得登錄序列號。從NCBI下載國內外有關短體線蟲種類和群體的rDNA 18S、28S D2-D3以及mtDNA-COI序列,利用軟件ClustalX1.81對所測短體線蟲序列和下載的短體線蟲序列進行比對分析。采用軟件MEGA4.0的鄰接法(neighbor-joining method)構建短體線蟲的rDNA 18S、28S D2-D3和mtDNA-COI序列系統進化樹,采用Boostrap值檢驗分支聚類的可靠性[30],進化樹顯示置信度>50%的數值。此外,使用MEGA4.0對鑒定的短體線蟲種類與不同地理種群進行分子序列的差異度和相似度分析。
1.5 SCAR-PCR特異性引物驗證
根據系統發育樹及序列比對分析結果,利用種特異性引物3對[18-20](表2)對不同DNA樣本進行擴增,PCR反應體系:DNA模板1 μL,5 μmol·L-1的上游引物和下游引物各2 μL,2×Ex Taq Mix 12.5 μL,ddH2O補足至25 μL。擴增條件:94℃預變性4 min;94℃變性30 s,60—66℃(按引物不同)退火30 s,72℃延伸45 s,共40個循環;72℃延伸10 min。PCR產物在1.2%瓊脂糖凝膠上電泳檢測,根據相應引物的擴增片段大小檢測線蟲的種類。
2 結果
2.1 rDNA與mtDNA通用引物擴增片段序列分析
首先用引物G18SU/R18TYL1擴增各線蟲的rDNA 18S區,均得到一條約900 bp的條帶(圖1-A),各片段經克隆測序后獲得的片段大小范圍在857—935 bp,序列比對分析顯示部分樣本間序列差異明顯,初步判斷小麥短體線蟲10個樣品中,有6個為短體線蟲混合種群侵染樣品,其余可能為短體線蟲單一種群侵染樣品。
根據18S序列分析比對結果,從6個混合種群侵染樣品中各選取序列有明顯差異的線蟲DNA樣本2個,從4個單一種群侵染樣品中各選取序列一致的線蟲DNA樣本2個(線蟲種群代碼見表1),用引物D2A/D3B擴增rDNA 28S D2-D3區,所有樣本均得到一條約780 bp的條帶(圖1-B),測序得到的片段大小范圍在771—784 bp;用引物JB3/JB5擴增mtDNA-COI區,均獲得了一條約400 bp的條帶(圖1-C),測序得到的片段大小范圍在415—417 bp。
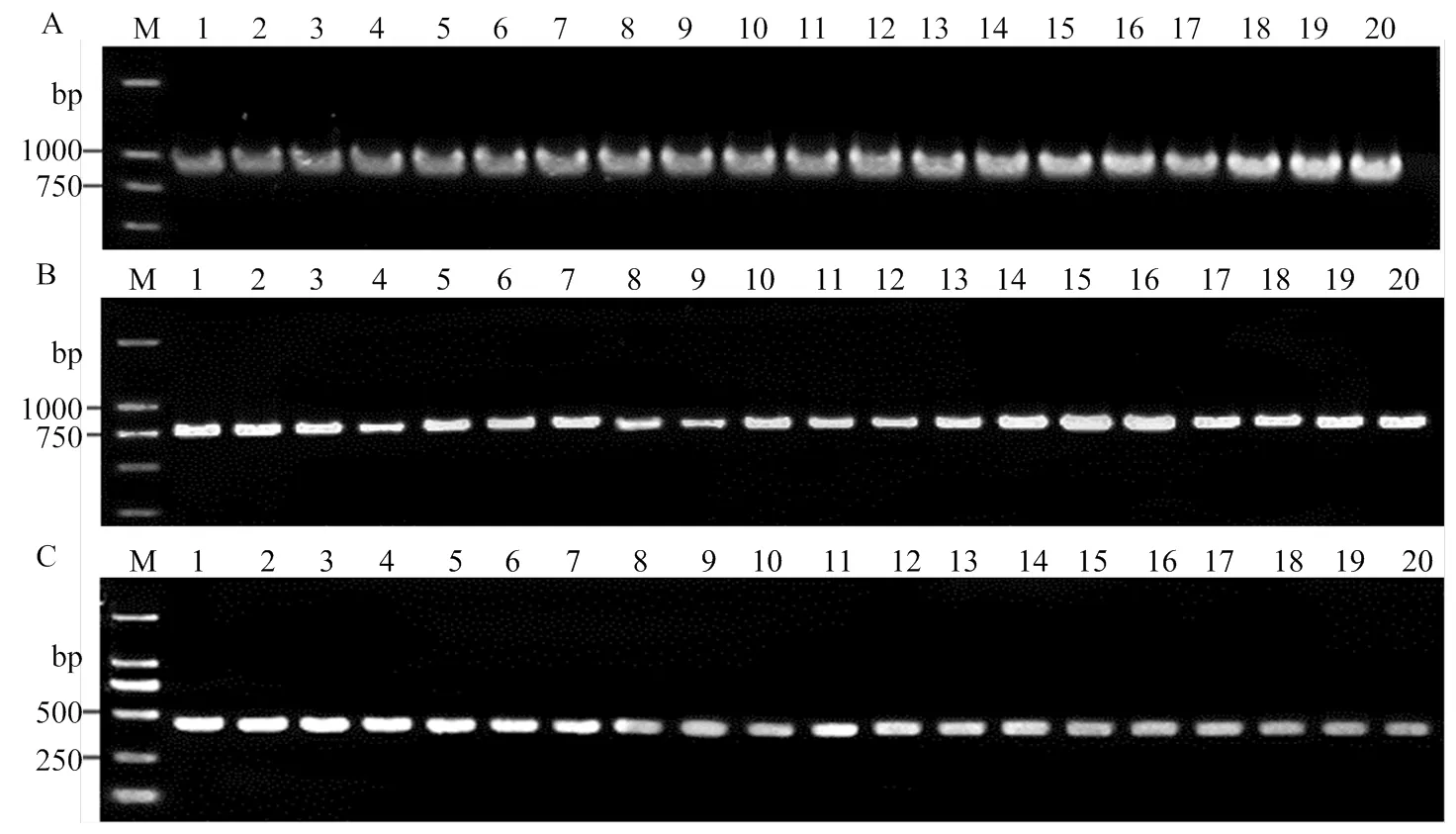
A: rDNA 18S; B: rDNA 28S D2-D3; C: mtDNA-COI.1-20: JS1-1, JS1-2, JS2-1, JS2-2, AH2-1, AH2-2, AH3-1, AH3-2, AH4-1, AH4-2, AH5-1, AH5-2, HN1-1, HN1-2, HN2-1, HN2-2, HN3-1, HN3-2, SD1-1, SD1-2; M: DNA marker DL2000
2.2 短體線蟲種群的系統進化分析
從NCBI分別下載來自美國、英國、土耳其、日本、荷蘭、比利時、澳大利亞、伊朗、越南和肯尼亞等國家的短體線蟲不同種群的rDNA 18S、28S D2-D3和mtDNA-COI序列,與本研究獲得的序列一起構建鄰接法系統進化樹(圖2—圖4),用長尾刺線蟲()和細刺線蟲()作為rDNA 18S樹的外群,用長尾刺線蟲作為rDNA28S D2-D3樹的外群,用相似穿孔線蟲()作為mtDNA-COI樹的外群。
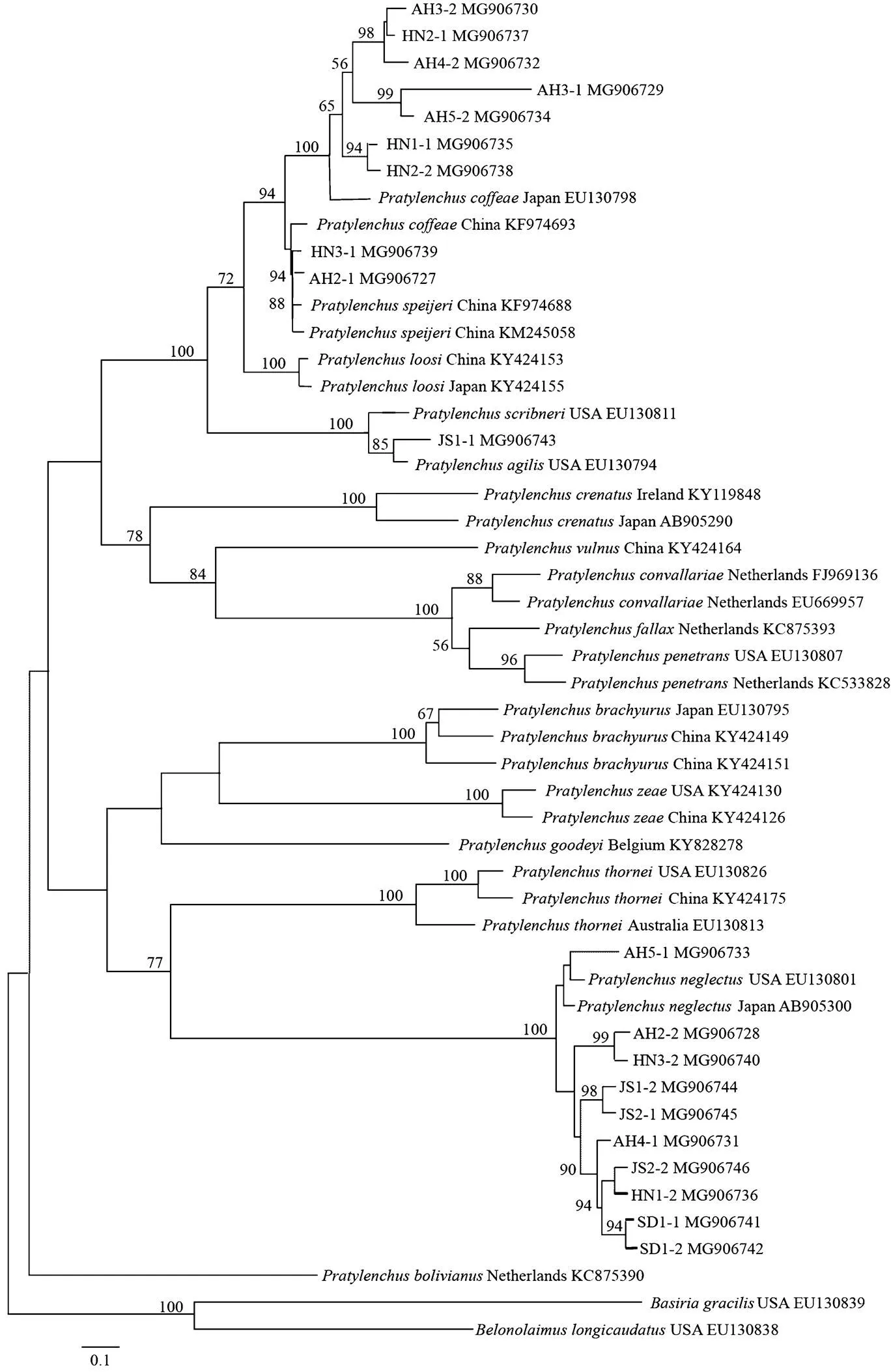
圖2 基于rDNA 18S序列構建的黃淮麥區短體線蟲種群鄰接法系統進化樹

圖3 基于rDNA 28S D2-D3序列構建的黃淮麥區短體線蟲種群鄰接法系統進化樹
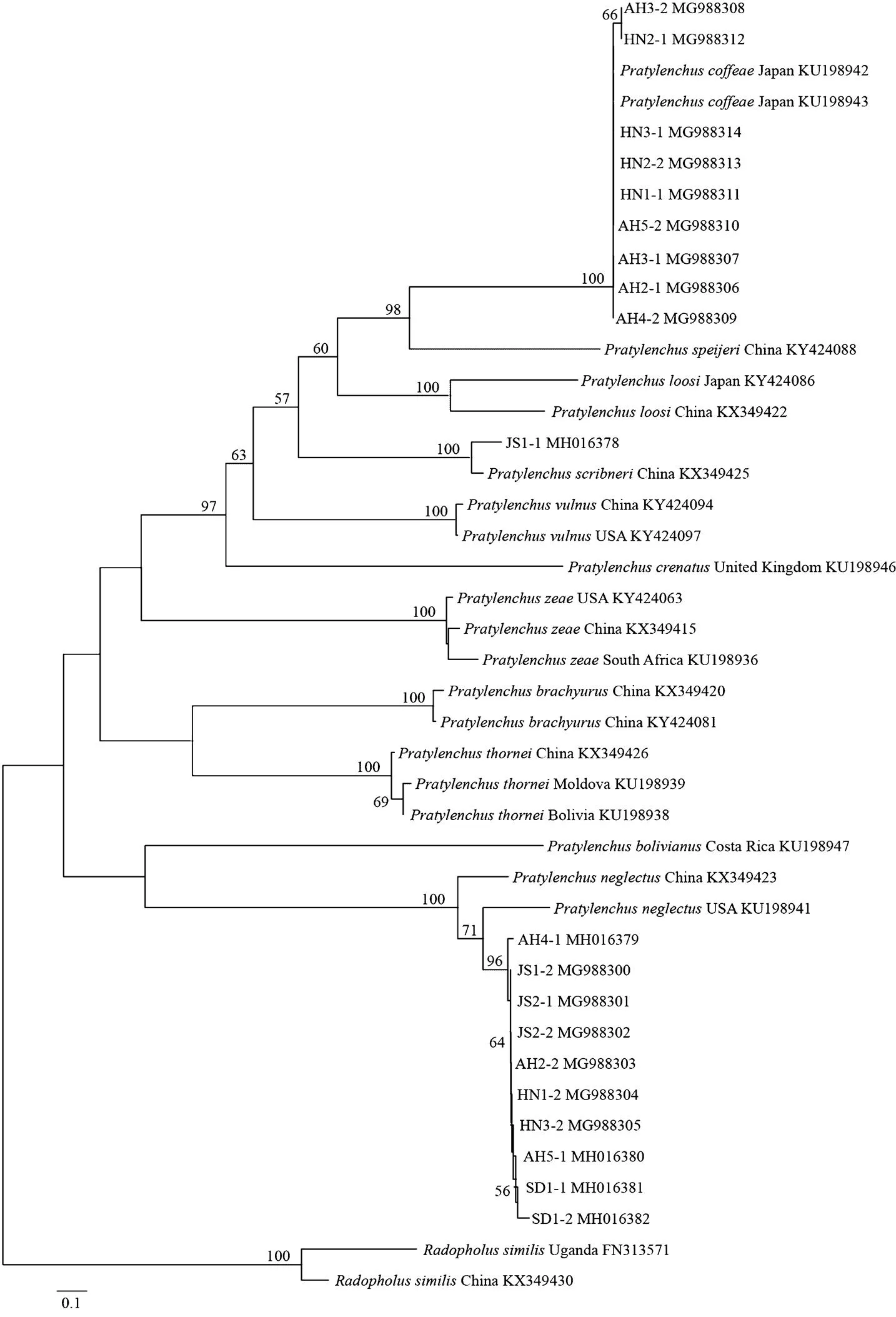
圖4 基于mtDNA-COI序列構建的黃淮麥區短體線蟲種群鄰接法系統進化樹
從rDNA 18S系統進化樹(圖2)可以看出,安徽小麥短體線蟲種群AH2-1、AH3-1、AH3-2、AH4-2和AH5-2,以及河南小麥的短體線蟲種群HN1-1、HN2-1、HN2-2和HN3-1,均與咖啡短體線蟲日本、中國群體和史佩奇短體線蟲()中國群體聚類在一個分支,置信度達到94%。安徽小麥短體線蟲種群AH2-2、AH4-1和AH5-1,江蘇小麥短體線蟲種群JS1-2、JS2-1和JS2-2,河南小麥短體線蟲種群HN1-2和HN3-2,以及山東小麥短體線蟲種群SD1-1和SD1-2,均與落選短體線蟲日本群體和美國群體聚類在一個大分支,置信度達到100%。而江蘇小麥短體線蟲種群JS1-1與斯克里布納短體線蟲美國群體和敏捷短體線蟲聚類于一個大分支,置信度達到100%。
基于rDNA 28S D2-D3序列構建的系統進化樹(圖3)顯示,AH2-1、AH3-1、AH3-2、AH4-2、AH5-2、HN1-1、HN2-1、HN2-2和HN3-1這9個種群均與咖啡短體線蟲的美國、伊朗、越南群體聚類在一個分支,置信度達到99%,且與史佩奇短體線蟲處于完全不同的進化分支;AH2-2、AH4-1、AH5-1、JS1-2、JS2-1、JS2-2、HN1-2、HN3-2、SD1-1和SD1-2這10個種群均與落選短體線蟲的中國、美國、加拿大群體聚類在一個分支,置信度達到100%;而種群JS1-1與斯克里布納短體線蟲的中國、美國群體和六裂短體線蟲的土耳其、比利時群體聚類在一個分支,置信度達到100%。
基于mtDNA-COI序列構建的系統進化樹(圖4)與rDNA 28S D2-D3進化樹樹形相似,20個短體線蟲種群分別聚類在咖啡短體線蟲、落選短體線蟲和斯克里布納短體線蟲的不同分支里,置信度均為100%。
2.3 短體線蟲種群的rDNA 28S和mtDNA-COI序列相似度分析
對咖啡短體線蟲安徽群體AH2-1、AH3-1、AH3-2、AH4-2和AH5-2,以及河南群體HN1-1、HN2-1、HN2-2和HN3-1的28S D2-D3序列進行比對分析,結果顯示,9個群體的序列差異值在0.000—0.010,相似度為99.0%—100%,它們與已知中國群體的差異值也在0.000—0.010,相似度為99.0%—100%,而與咖啡短體線蟲的巴西、美國、日本、越南群體的序列差異值在0.000—0.013,相似度為98.7%—100%(表3);此外,這9個群體的mtDNA-COI序列差異值在0.000—0.003,相似度為99.7%—100%,它們與咖啡短體線蟲日本群體(KU1098942和KU198943)的序列差異值同樣是在0.000—0.003,相似度為99.7%— 100%(表4)。

表3 咖啡短體線蟲不同群體間的rDNA 28S D2-D3 序列差異值
對落選短體線蟲安徽群體AH2-2、AH4-1和AH5-1,江蘇群體JS1-2、JS2-1和JS2-2,河南群體HN1-2和HN3-2,以及山東群體SD1-1和SD1-2的28S D2-D3序列進行相似度分析,從表5中可以看出,這10個群體序列間的差異值在0.000—0.020,相似度在98.0%—100%;它們與已知中國群體的差異值在0.011—0.020,相似度為98.0%—98.9%,而與落選短體線蟲美國、比利時、伊朗、韓國、英國、捷克、玻利維亞群體的序列差異值同樣是在0.000—0.020,相似度為98.0%—100%。此外,這10個群體的mtDNA-COI序列差異值在0.000—0.003之間,相似度在99.7%—100%,它們與落選短體線蟲美國群體(KU198941)和中國群體(KY424103)的序列差異值在0.029—0.032,相似度在96.8%—97.1%(表6)。
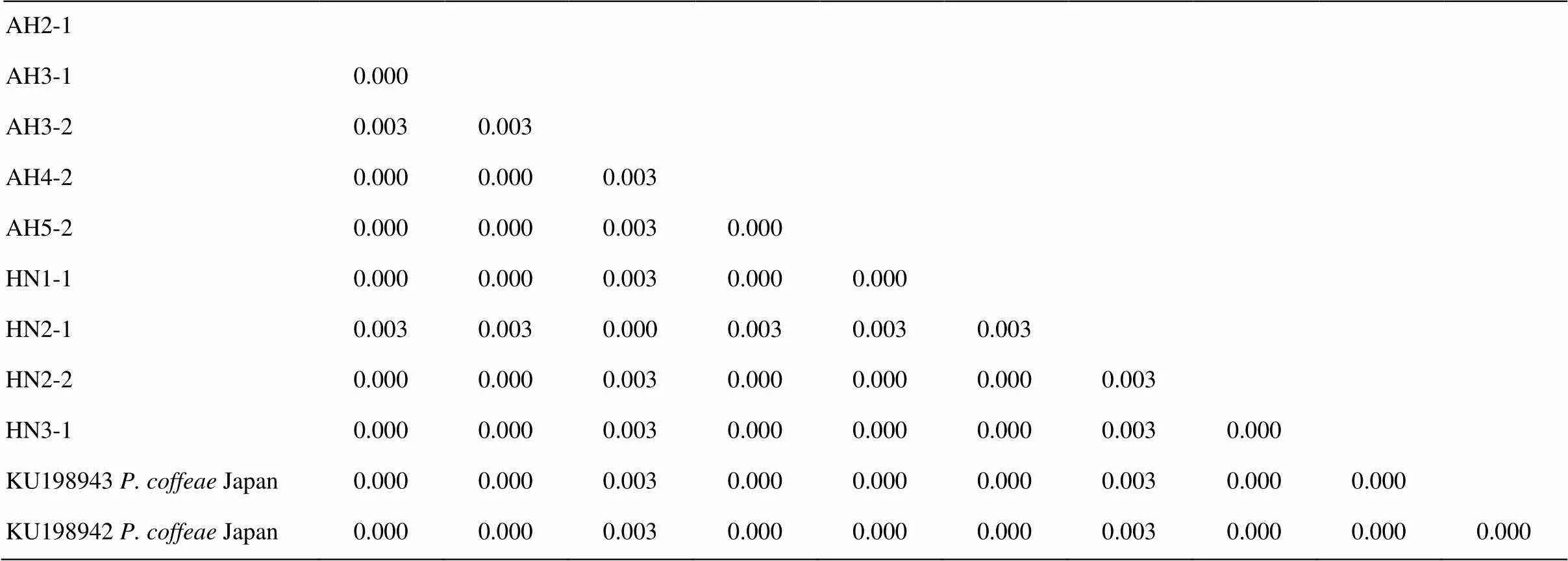
表4 咖啡短體線蟲不同群體間的mtDNA-COI序列差異值

表5 不同落選短體線蟲群體間的rDNA 28S D2-D3序列差異值

表6 不同落選短體線蟲群體間的mtDNA-COI序列差異值
將江蘇群體JS1-1的28S D2-D3序列與斯克里布納短體線蟲美國群體(KT873859、KX842628和EU130865)以及中國群體(KM094196和JX047004)的序列進行分析,結果顯示它們之間的差異值在0.000—0.004,相似度為99.6%—100%;JS1-1的mtDNA-COI序列與斯克里布納短體線蟲中國群體(KX349425)的序列差異值為0.012,相似度為98.8%。
通過對黃淮流域4省10個小麥短體線蟲樣品的種群系統進化樹及序列相似性的綜合分析,揭示江蘇沛縣樣品JS2和山東濰坊樣品SD1是落選短體線蟲單一種群,河南永城樣品HN2和安徽淮北樣品AH3是咖啡短體線蟲單一種群,江蘇徐州樣品JS1是落選短體線蟲和斯克里布納短體線蟲的混合種群,而安徽蕭縣樣品AH2和AH5、安徽淮北樣品AH4以及河南永城樣品HN1和HN3,均為咖啡短體線蟲和落選短體線蟲的混合種群(表1)。
2.4 短體線蟲種群的SCAR引物檢測
利用SCAR引物對表1中的20個短體線蟲種群DNA樣本進行擴增,結果顯示,咖啡短體線蟲SCAR引物對PC1/PC2僅從AH2-1、AH3-1、AH3-2、AH4-2、AH5-2、HN1-1、HN2-1、HN2-2和HN3-1等9個樣本中各擴增出一條約630 bp的條帶(圖5條帶7、10、13所示),與預期的條帶大小相一致,而落選短體線蟲SCAR引物對PNEG-F1/D3B5和斯克里布納短體線蟲SCAR引物對PsF7/PsR7從上述樣品中均沒有擴增出條帶,表明這些樣本均為咖啡短體線蟲。引物對PNEG-F1/D3B5僅從JS1-2、JS2-1、JS2-2、AH2-2、AH4-1、AH5-1、HN1-2、HN3-2、SD1-1和SD1-2等10個樣本中各擴增出約一條約140 bp的條帶(圖5條帶5、17、20所示),與預期的落選短體線蟲擴增片段大小相一致;而引物對PsF7/ PsR7僅從JS1-1樣本中擴增出130 bp的條帶,與預期的斯克里布納短體線蟲擴增片段大小相一致(圖5條帶3所示)。SCAR檢測結果表明,10個小麥短體線蟲樣品里20個短體線蟲種群的單條DNA樣本中,有9個為咖啡短體線蟲,10個為落選短體線蟲,僅1個為斯克里布納短體線蟲,這與上述基于rDNA和mtDNA序列分析得出的結果相一致。
3 討論
短體線蟲屬不僅種類多,而且種間形態特征差異較小,種類鑒定難度比較大;此外,線蟲的形態特征測計往往需要一定數量的成蟲,對于群體密度較低或者混合種群的樣本而言,形態測計容易產生誤差,會導致依據形態特征鑒定的種類與實際情況不符。而基于線蟲DNA發展起來的多種分子鑒定技術,作為形態鑒定的一種輔助手段已經廣泛應用于線蟲近似種的分類鑒定中[31]。盡管目前已開發了針對多種短體線蟲的SCAR特異性引物[18-21],但是采用該方法需要分別使用多種引物對同一DNA樣本進行擴增,如果特異性引物與樣本的種類沒有對應,就不會有擴增條帶產生,從而無法對線蟲種類進行檢測鑒定。本文首先通過擴增短體線蟲的rDNA 18S、28S D2-D3和mtDNA-COI片段,利用所測得的序列構建系統進化樹并進行序列相似度分析,初步明確樣本的種類,并在此基礎上應用SCAR特異性引物進行PCR驗證,成功地利用單條短體線蟲DNA樣本對采自黃淮流域4省的10個小麥短體線蟲樣品進行了種類鑒定。
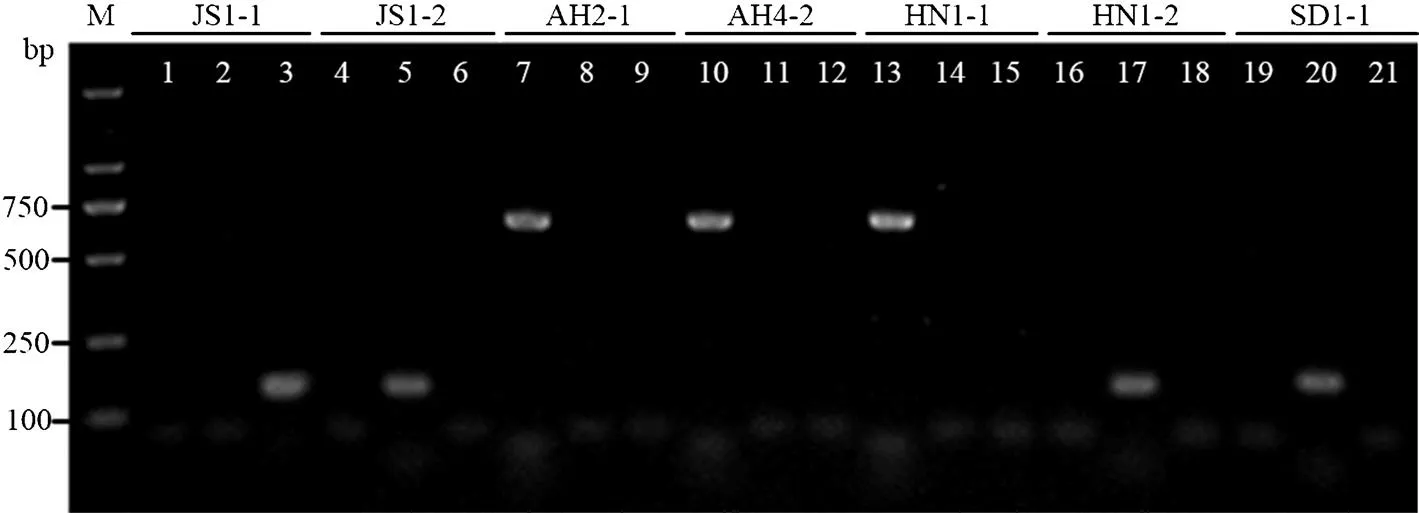
1、4、7、10、13、16、19:咖啡短體線蟲SCAR引物PC1/PC2 SCAR primers PC1/PC2 for P. coffeae;2、5、8、11、14、17、20:落選短體線蟲SCAR引物PNEG-F1/D3B5 SCAR primers PNEG-F1/D3B5 for P. neglectus;3、6、9、12、15、18、21:斯克里布納短體線蟲SCAR引物PsF7/ PsR7 SCAR primers PsF7/ PsR7 for P. scribneri;M:DNA marker DL2000
通過分析rDNA 18S系統進化樹筆者發現,本研究測定的咖啡短體線蟲群體與史佩奇短體線蟲群體聚類在一個進化分支中,而在rDNA 28S和mtDNA-COI樹中,咖啡短體線蟲群體與史佩奇短體線蟲群體聚類在不同的進化分支中。究其原因可能是由于rDNA 18S基因進化速率相對較慢,種間保守性高,更適合于屬間系統進化分析[32],因此無法將咖啡短體線蟲和史佩奇短體線蟲區分開;rDNA 28S基因是區分動物物種及其近緣種的有效標記之一[33],在植物寄生線蟲同源性比較和系統進化研究中也有廣泛應用;mtDNA-COI基因由于具有單親遺傳、無重組、進化速率相對較快等特點,在分子遺傳學研究中發揮著重要作用[34],也是近年來物種條形碼研究的熱點。通過進一步分析咖啡短體線蟲與落選短體線蟲rDNA 28S和mtDNA-COI序列可以看出,后者在兩種短體線蟲的種內遺傳變異較前者更小,物種的辨識度更高。JansseN等[4]也證實mtDNA-COI分子靶標相比較rDNA 28S和ITS靶標可以更好地將穿刺短體線蟲復合種群(species complex)成員穿刺短體線蟲、偽短體線蟲和鈴蘭短體線蟲有效區分開,因此本文的結果也支持了mtDNA-COI分子靶標比rDNA 18S和28S分子靶標更適合于短體線蟲種類的分子鑒定和系統進化研究。
李廣帥[35]對河南省9個地區采集的23個小麥短體線蟲樣品進行了形態學和分子鑒定,認為其中有15個樣品為敏捷短體線蟲,其余8個樣品為盧斯短體線蟲。本研究對河南商丘地區采集的樣品進行了鑒定,并未發現上述兩種短體線蟲,由于采樣地點不同,無法排除不同地域線蟲種類存在差異的可能性,但通過進一步的序列比對分析,筆者發現河南省鄭州市惠濟區敏捷短體線蟲群體ZMZZ的ITS序列(JQ039330)[36]與已報道的敏捷短體線蟲美國群體(FJ712889)及中國山西群體(KC952982)[37]的相似性僅為90.8%和91.0%,而中國山西群體與美國群體的相似性高達98.0%。目前的有效種地位受到質疑,被認為可能是的同種異名[38-39],本研究分析發現ZMZZ群體與斯克里布納短體線蟲江蘇沛縣群體JS1-1(MG906767)和美國群體(KX842626)的ITS序列相似性僅為88.9%和90.0%,由于ZMZZ群體僅報道了ITS序列,無法進一步比較分析,因此,河南ZMZZ群體的種類還有待核實與商榷。
此外,盧斯短體線蟲是咖啡短體線蟲復合種群(species complex)成員之一[40],形態特征與咖啡短體線蟲非常相似,盧斯短體線蟲區別于咖啡短體線蟲的形態特征僅僅為更細的蟲體,陰門位置稍偏后,尾形偏窄,尾末端窄圓或鈍尖[3]。通過文獻核查,筆者發現此前報道的盧斯短體線蟲8個河南群體的尾形,明顯寬于參考文獻描繪的盧斯短體線蟲尾形,且尾末端寬圓,由于這8個河南群體缺乏清晰的分子信息,因此這些河南群體的種類還有待進一步的驗證。
4 結論
通過rDNA 18S、28S以及mtDNA-COI序列分析和SCAR鑒定方法,對我國黃淮流域江蘇、安徽、河南、山東4省小麥孢囊線蟲發病田中的10個短體線蟲樣品進行了種類鑒定,揭示黃淮麥區的短體線蟲種類有咖啡短體線蟲、落選短體線蟲和斯克里布納短體線蟲,其中落選短體線蟲是優勢種群,短體線蟲不同種群復合侵染小麥發生比較普遍。基于mtDNA-COI基因構建的系統進化樹可以有效區分短體線蟲的近緣種,相比rDNA 18S和28S基因更適于短體線蟲種類的分子鑒定。
[1] 顧建鋒, 王江嶺, 何潔,陳先鋒. 4種常見短體線蟲形態特征比較鑒別. 植物檢疫, 2013, 27(6): 69-71.
Gu J F, Wang J L, He J, Chen X F. Morphological characters of four commonspecies., 2013, 27(6): 69-71. (in Chinese)
[2] 趙立榮, 何日榮, 武目濤,胡學難, 王衛芳.3種截獲短體線蟲的形態與分子特征研究. 植物檢疫, 2014, 28(4): 50-54.
Zhao L R, He R R, Wu M T, Hu X N, Wang W F. Morphological and molecular characterization of threenematodes imported from Japan., 2014, 28(4): 50-54. (in Chinese)
[3] Geraert E.. Gent. Belgium: Academia Press, 2013.
[4] Janssen T, Karssen G, Orlando V, Subbotin S A, Bert W. Molecular characterization and species delimiting of plant-parasitic nematodes of the genusfrom thegroup (Nematoda: Pratylenchidae)., 2017, 117: 30-48.
[5] 李廣帥, 施艷, 崔娟, 杜鵑, 邢小萍, 王振躍. 小麥根腐線蟲發生規律及其對小麥產量的影響. 河南農業科學, 2011, 40(4): 93-97.
Li G S, Shi Y, Cui J, Du J, XinG X P, Wang Z Y. Occurrence law of wheat root lesion nematode and its impact on wheat yield., 2011, 40(4): 93-97. (in Chinese)
[6] 王暄, 孫成剛, 方亦午, 向桂林, 劉炳良, 宋志強, 高菲菲, 李紅梅. 基于GIS的小麥孢囊線蟲病在江蘇省的發生分布與群體密度分析. 植物病理學報, 2012, 42(5): 515-524.
Wang X, Sun C G, Fang Y W, Xiang G L, Liu B L, Song Z Q, Gao F F, Li H M. Analyses of distribution and population density of cereal cyst nematodes on wheat in Jiangsu Province based on GIS., 2012, 42(5): 515-524. (in Chinese)
[7] 劉榮榮, 王暄, 李紅梅,王珍, 劉海璐. 河南省小麥主產區菲利普孢囊線蟲與禾谷孢囊線蟲發生情況調查.植物保護, 2017, 43(5): 157-163.
Liu R R, Wang X, Li H M, Wang Z, Liu H L. Survey of the occurrence ofandon wheat in main growing area in Henan Province., 2017, 43(5): 157-163. (in Chinese)
[8] Nicol J M, Rivoal R, Taylor S, Zaharieva M. Global importance of cyst (spp.) and lesion nematodes (spp.) on cereals: yield loss, population dynamics, use of host resistance and integration of molecular tools., 2003, 2: 1-19.
[9] 王振躍, 崔娟, 杜鵑, 李洪連, 崔長富, 崔麗娜. 禾谷類作物根腐線蟲病研究進展. 河南農業科學, 2009, 38(12): 18-21, 25.
Wang Z Y, Cui J, Du J, Li H L, Cui C F, Cui L N. Advance in root lesion nematodes on cereal crops., 2009, 38(12): 18-21, 25. (in Chinese)
[10] 楊榮錚, 陳建軍, 梁文慧, 陶衛平. 安徽省午季作物寄生線蟲種類的調查. 安徽農業大學學報, 1983, 10(1): 91-96.
Yang R Z, Chen J J, Liang W H, Tao W P. Investigation of parasitic nematode species on summer crops in Anhui Province., 1983, 10(1): 91-96. (in Chinese)
[11] 李篤肇. 四川省植物寄生短體線蟲種的記述. 西南農學院學報, 1985, 7(2): 49-51.
Li D Z. Descriptions of some species of parasitic nematodes of genuson plant roots in Sichuan Province., 1985, 7(2): 49-51. (in Chinese)
[12] 方羽生, 尹淦鏐. 植物病原線蟲短體屬種類的研究. 華南師范大學學報(自然科學版), 1994(4): 32-41.
Fang Y S, YiN G L. Study on pathogenetic nematodes (: Pratylenchinae) from roots of crops in Guangxi Zhuang Autonomous Region., 1994(4): 32-41. (in Chinese)
[13] 吳慧平, 楊傳廣, 陳良宏, 檀根甲, 王向陽. 安徽省小麥根際線蟲的鑒定和分布. 安徽農業大學學報, 2010, 37(2): 189-195.
Wu H P, Yang C G, Chen L H, Tan G J, Wang X Y. Identification and distribution of rhizosphere nematodes of wheat in Anhui Province., 2010, 37(2): 189-195. (in Chinese)
[14] 王振躍, 施艷, 李廣帥, 崔娟, 杜鵑, 李洪連. 河南省小麥根腐線蟲病的發生危害及病原鑒定. 植物保護學報, 2012, 39(4): 297-302.
Wang Z Y, Shi Y, Li G S, Cui J, Du J, Li H L. Occurrence, damage and pathogen identification of wheat root lesion nematode disease in Henan Province., 2012, 39(4): 297-302. (in Chinese)
[15] 于焦, 金惺惺, 秦萌, 吳文佳, 徐春玲, 謝輝.西藏農作物短體線蟲種類的鑒定和描述. 華中農業大學學報, 2017, 36(5): 20-24.
Yu J, Jin X X, Qin M, Wu W J, Xu C L, Xie H. Identification and description of three species of genusfrom crops in Tibet., 2017, 36(5): 20-24. (in Chinese)
[16] Pinochet P, Cenis J L, Fernández C, Doucet M, Maruli J. Reproductive fitness and random amplified polymorphic DNA variation among isolates of., 1994, 26(3): 271-277.
[17] Ouri Y, Mizukubo T. Discrimination of sevenspecies (Nematoda: Pratylenchidae) in Japan by PCR-RFLP analysis., 1999, 34(2): 205-211.
[18] Uehara T, Mizukubo T, Kushida A, Momota Y. Identification ofandusing specific primers of PCR amplification of ribosomal DNA., 1998, 44(4): 357-368.
[19] Yan G P, Smiley R W, Okubara P A, Skantar A, Easley S A, Sheedy J G, Thompson A L. Detection and discrimination ofandin DNA extracts from soil., 2008, 92(11): 1480-1487.
[20] Huang D Q, Yan G P. Specific detection of the root-lesion nematodeusing conventional and real-time PCR., 2017, 101(2): 359-365.
[21] Al-Banna L, Ploeg A T, Williamson V M, Kaloshian I. Discrimination of sixspecies using PCR and species- specific primers., 2004, 36(2): 142-146.
[22] LiuX T, Wang H H, Lin B R, Tao Y, Zhuo K, Liao J L. Loop-mediated isothermal amplification based on the mitochondrialregion to detect,2017, 148(2): 435-446.
[23] 魏亞東, 容萬韜, 趙立榮, 王金成, 黃國明, 郭京澤, 孫建華. 五種短體線蟲DNA條形碼鑒定方法. 華北農學報, 2013, 28(6): 136-139.
Wei Y D, Rong W T, Zhao L R, Wang J C, Huang G M, Guo J Z, Sun J H. Identification of 5species utilizing DNA barcoding method., 2013, 28(6): 136-139.(in Chinese)
[24] Subbotin S A, Ragsdalea E J, Mullens T, Roberts P A, Mundo-Ocampo M, Baldwin J G. A phylogenetic framework for root lesion nematodes of the genus(Nematoda): evidence from 18S and D2-D3 expansion segments of 28S ribosomal RNA genes and morphological characters., 2008, 48(2): 491-505.
[25] 王金成, 魏亞東, 顧建鋒, 張瑞豐, 黃國明, 王暄, 李紅梅, 孫建華. 基于核糖體ITS區和28S rRNA D2-D3區的短體線蟲系統發育研究. 動物分類學報, 2012, 37(4): 687-693.
Wang J C, Wei Y D, Gu J F, Zhang R F, Huang G M, Wang X, Li H M, Sun J H. Phylogenetic analysis of(Nematoda, Pratylenchidae) based on ribosomal internal transcribed spacers (ITS) and D2-D3 expansion segments of 28S rRNA gene., 2012, 37(4): 687-693. (in Chinese)
[26] 宋志強, 王暄, 林宇, 遲元凱, 鞠玉亮, 李紅梅. 土壤中南方根結線蟲的實時熒光PCR檢測和定量. 植物保護學報, 2013, 40(3): 255-260.
Song Z Q, Wang X, Lin Y, Chi Y K, Ju Y L, Li H M. Detection and quantification ofin soil sample using real-time PCR., 2013, 40(3): 255-260. (in Chinese)
[27] Chizhov V N, Chumakova O A, Subbotin S A, Baldwin J G. Morphological and molecular characterization of foliar nematodes of the genus:and(Nematoda: Aphelenchoididae) from the Main Botanical Garden of the Russian Academy of Sciences, Moscow.,2006, 14(2): 179-184.
[28] Subbotin S A, Sturhan D, Chizhov V N, Vovlas N, Baldwin J G. Phylogenetic analysis of Tylenchida Thorne, 1949 as inferred from D2 and D3 expansion fragments of the 28S rRNA gene sequences.,2006, 8(3): 455-474.
[29] Derycke S, Vanaverbeke J, Rigaux A, Backeljau T, Moens T. Exploring the use of cytochrome oxidase c subunit I (COI) for DNA barcoding of free-living marine nematodes., 2010, 5(10): e13716.
[30] Saitou N, Nei M. The neighbor-joining method, a new method for reconstructing phylogenetic trees., 1987, 4(4): 406-425.
[31] 王寧, 顧建鋒, 王暄, 李紅梅. 進境雞爪槭中日本短體線蟲的鑒定. 南京農業大學學報, 2014, 37(4): 76-82.
Wang N, Gu J F, Wang X, Li H M. Identification ofintercepted infrom Japan., 2014, 37(4): 76-82. (in Chinese)
[32] Holterman M,van der Wurff A,van den Elsen S,van Megen H,Bongers T,Holovachov O,Bakker J,Helder J.Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown clades., 2006, 23(9): 1792-1800.
[33] Sonnenberg R, Nolte A W, Tautz D. An evaluation of LSU rDNA D1-D2 sequences for their use in species identification., 2007, 4: 6.
[34] Hebert P D, Ratnasingham S, de Waard J R. Barcoding animal life: cytochrome c oxidase subunit I divergences among closely related species., 2003, 270 (Suppl. 1): S96-99.
[35] 李廣帥. 河南省小麥根腐線蟲病原鑒定和RAPD分析[D]. 鄭州: 河南農業大學, 2011.
Li G S. The identification and RAPD analysis of wheat root lesion pathogens in Henan Province[D]. Zhengzhou: Henan Agricultural University, 2011. (in Chinese)
[36] Wang Z Y, Shi Y, Li H L, Zhang M. First report of the lesion nematode,, parasitizing wheat in China., 2012, 96(5): 773.
[37] Shi H L, Zheng J W. Morphological and molecular identification ofassociated with wheat roots from Shanxi, China., 2012, 44(4): 491.
[38] Hernández M, Jordana R, Goldaracena A, Pinochet J. SEM observations of nine species of the genusFilipjev, 1936 (Nematoda: Pratylenchidae)., 2000, 3(2): 165-174.
[39] Castillo P, Vovlas N.(). Leiden, The Netherlands: Brill Academic Publishing, 2007.
[40] De Luca F, Troccoli A, Duncan L W, Subbotin S A, Waeyenberge L, Coyne D L, Brentu F C, Inserra R N.n. sp. (Nematoda: Pratylenchidae), a new root-lesion nematode pest of plantain in West Africa., 2012, 14(8): 987-1004.
(責任編輯 岳梅)
Molecular Identification ofSpecies in 10 Samples Collected from Wheat Field in Huanghuai Region of China
Liu HaiLu, Wang Xuan, Li Hongmei, Li Yanxia, Xue Bowen, Ma Jukui
(College of Plan Protection, Nanjing Agricultural University/Key Laboratory of Integrated Management of Crop Diseases and Pests, Ministry of Education, Nanjing 210095)
【Objective】Thespecies are migratory endoparasites of plant roots, causing root lesion of many crops and great damage of agricultural production all over the world. In order to clarify the species of genusco-infection withon wheat from Huanghuai region of China, 10 samples were collected from wheat field in 4 provinces of the region and thespecies were molecularly identified. The phylogenetic relationship ofspecies and genetic variation of intraspecific populations were analyzed. The results will provide valuable information for the integrated control of nematode diseases on wheat root. 【Method】Thenematodes were extracted from 10 samples collected from wheat fields infected within Jiangsu, Anhui, Henan and Shandong provinces. Fivenematodes were randomly picked up from each sample, and the DNA of individual nematode was extracted as the template for PCR amplification. The fragments of rDNA 18S region were amplified and the PCR products were sequenced. The BLAST alignment of 18S sequences revealed the differentspecies may present in these samples. The DNA templates of two representative specimens from each sample were selected for the further amplification of fragments from rDNA 28S D2-D3 region and mtDNA-COI gene. The fragments were sequenced and the BLAST alignments were performed. The phylogenetic trees were constructed on basis of rDNA 18S, 28S D2-D3 and mtDNA-COI sequences using neighbor-joining method by MEGA4.0 software, respectively. Thespecies identification was supported by the analyses of phylogenetic relationships and sequence similarities. The species-specific primers as the sequence-characterized amplified regions (SCAR) markers were used to validate the identification. 【Result】The fragments amplified from rDNA 18S region of 50 individual nematodes were sequenced and the sizes were 857-935 bp. The BLAST searching revealed that the mixedpopulations might present in some samples. The sequence sizes of the rDNA 28S D2-D3 fragments amplified from 20 selected specimens were 771-784 bp, the sizes of mtDNA-COI were 415-417 bp. The phylogenetic analyses as well as the comparisons of sequence similarities both demonstrated that threespecies including,and,were found in 10 samples collected from wheat fields in 4 provinces of Huanghuai region. Among 10 samples, the sample JS2 from Peixian of Jiangsu Province and SD1 from Weifang of Shandong Province were infected only with, HN2 from Yongcheng of Henan Province and AH3 from Huaibei of Anhui Province were infected only with. The samples AH2 and AH5 from Xiaoxian and AH4 from Huaibei of Anhui Province as well as HN1 and HN3 from Yongcheng of Henan Province were all co-infected withand, and JS1 from Xuzhou of Jiangsu Province was co-infected with. The DNA templates from 20 representative specimens were amplified using SCAR primers. The results showed that a single band of 140 bp was amplified from JS1-2, JS2-1, JS2-2, AH2-2, AH4-1, AH5-1, HN1-2, HN3-2, SD1-1 and SD1-2 using specific primer PNEG-F1/D3B5 for, a single band of 630 bp was amplified from AH2-1, AH3-1, AH3-2, AH4-2, AH5-2, HN1-1, HN2-1, HN2-2 and HN3-1 using primer PC1/PC2 for,a single band of 130 bp was amplified from JS1-1 using primer PsF7/PsR7 forThe results of SCAR detection confirmed the species identification mentioned above.【Conclusion】The molecular identification demonstrated that three species,,and, were found in wheat fields infested withfrom four provinces in Huanghuai region, andis the dominant species. The co-infection of differentspecies occurred quiet common in wheat field from the region. The phylogenetic tree based on mtDNA-COI gene can effectively distinguish the close relatedspecies, therefore, it is more suitable to identifyspecies than rDNA 18S and 28S markers.
spp.; species identification; rDNA; mtDNA; phylogeny; SCAR
2018-02-05;
2018-05-02
國家公益性行業(農業)科研專項(201503114)、國家自然科學基金(31471751)
劉海璐,E-mail:2015102038@njau.edu.cn。通信作者王暄,E-mail:xuanwang@njau.edu.cn
10.3864/j.issn.0578-1752.2018.15.006
