青稞種質資源遺傳多樣性分析與核心種質群體的構建
2018-08-28 08:38:18原紅軍曾興權徐其君王玉林尼瑪扎西
麥類作物學報 2018年8期
原紅軍,曾興權,徐其君,王玉林,扎 桑,尼瑪扎西
(1.省部共建青稞和牦牛種質資源與遺傳改良國家重點實驗室, 西藏拉薩 850002; 2.西藏自治區農牧科學院 農業研究所,西藏拉薩 850002; 3.西藏自治區農牧研究院,西藏拉薩 850002)
青稞(HordeumvulgareL.) 作為大麥屬重要的農作物,廣泛分布于全球各地,如歐洲、美國和澳洲等地,在我國主要分布在西藏及其周邊地區。研究發現,西藏地區是世界栽培大麥的重要起源中心之一[1]。因其具有對高原極端氣候環境的適應性,在距今約4 000年前,青稞就已作為藏區人民重要的糧食作物得到廣泛種植。目前,青稞在西藏地區種植面積超過70%的耕作面積[2]。青稞的β-葡聚糖(β-glucan) 含量較高,有重要的營養價值和保健功能。
種質資源的遺傳多樣性和親緣關系評估,對特異種質的選擇和利用具有重要意義。在喜馬拉雅地區,青稞具有豐富的種質資源,已有許多關于喜馬拉雅地區的大麥資源遺傳多樣性的探究。在尼泊爾境內,不同青稞地方種質的農藝性狀和抗病性具有顯著的差異[3]。Pandey等[4]研究發現,青稞群體結構可能與資源的地理分布有關。已有研究所涉及資源群體較小,其結果可能具有一定的局限性。
尼瑪扎西等[2]對西藏青稞品種拉薩勾芒進行了全基因組測序,并繪制了青稞參考基因組草圖,這對青稞重要性狀QTL定位、基因挖掘以及青稞遺傳改良具有重要的意義。植物中常用的QTL定位方法主要是連鎖分析(linkage mapping)和關聯分析(association mapping),連鎖分析基于雙親分離群體,關聯分析則利用自然資源群體[5]。相比而言,全基因組關聯分析(genome-wide association study, GWAS)的功能更為強大和高效,在較低成本條件下,可實現對QTL的高精度解析[6]。基于自然資源群體的GWAS能將鑒定到的QTL通過分子標記輔助育種,利于篩選含優良等位基因的材料,加速青稞育種進程[7]。核心種質可以較小的群體代表絕大多數遺傳變異類型,是GWAS研究的重要內容之一。研究者已經利用核心自然群體對水稻[8-9]、玉米[10]、油菜[11]和花生[12]等重要農作物進行了QTL定位,發現了大量主效QTL和基因。
本研究擬利用95對SSR引物對1 220份來源廣泛的青稞種質材料進行基因型分析,分析其遺傳多樣性及親緣關系、群體結構及遺傳分化,構建一套合適的青稞核心種質,對其更有效利用和篩選優良親本提供參考。
1 材料與方法
1.1 試驗材料
以來自全球共計22個國家和地區的1 220份青稞為材料,其中,1 055份材料來自西藏,30份來自青海,28份來自四川等地,74份國外材料分別來自歐洲、加拿大和澳大利亞等地。所有材料均由中國農業科學院國家種質資源平臺提供。
1.2 試驗方法
1.2.1 SSR基因型分析
對每個種質材料采用修改的CTAB(Cetyltrimethylammonium bromide)法[13],隨機選擇一個單株的幼嫩葉片提取DNA。利用1%的瓊脂糖膠和lambda DNA,評估DNA的提取質量。利用本課題組前期開發的95對高質量SSR引物[14]對所有1 220份青稞材料進行基因型分析。PCR產物利用毛細管電泳(ABI3730 Genetic Analyzer Applied Biosystems)進行檢測。采用GeneMarker V2.1軟件鑒定SSR引物的不同等位變異。為了避免SSR同源片段的影響,本研究將SSR等位變異進行二進制編碼(1和0),記為該等位變異出現與否[15]。每對SSR引物的統計參數為不同等位變異的平均值。
1.2.2 遺傳多樣性分析
統計每對SSR引物的等位變異數目(allelic count)、基因多態性(Gene diversity)、多態性信息含量(Polymorphism information index, PIC) 和香農系數(Shannon index)。其中,變異數、基因多態性和PIC值采用PowerMarker V3.51軟件計算[16]。香農系數利用PopGene V1.31軟件包計算[17]。考慮群體大小對多樣性統計的影響,利用R程序包計算每個位點的等位變異豐度(allelic richness)。利用R程序中的t.test函數評估不同多樣性參數在亞群間的差異顯著性[18]。
1.2.3 群體結構和分化分析
采用STRUCTURE V2.2軟件[19]對1 220份青稞材料的群體結構進行分析。假定亞群數目K為1~10,每個K值進行5次模擬運算,每次模擬進行10 000次預迭代(length of burn-in period)和10 000次馬科夫鏈蒙特卡羅迭代(Markov chain monte carlo,MCMC),模型設定為混合和頻率相關模型。依據軟件輸出的后驗概率值[LnP(D)]和連續兩個LnP(D)間的變化速率(△K)[20]確定最合適的亞群數。計算每個品種源于不同亞群的概率,當某亞群對應值最大時[21],相應品種被劃分到該亞群。
利用R程序的prcomp函數對1 220份青稞材料進行主成分分析(principal component analysis, PCA),分析材料間的遺傳相似性。利用R程序的Chisq.test函數來評估群體結構和材料來源(geographic origin)的相關性。采用Arlequin V3.1[22]進行分子方差分析(analysis of molecular variance, AMOVA)和pairwiseFST分析,用其評估不同亞群間的遺傳分化程度,通過1 000 次排列檢驗來評估群體遺傳分化差異顯著性。
1.2.4 核心種質構建
利用R軟件包corehunter[23]從1 220個青稞材料中構建一套核心種質。軟件包同時考慮樣本間距離(average entry-to-nearest-entry distance)和基因多態性,最優核心種質將實現有效平衡等位變異多樣性和遺傳差異。通過500次蒙特卡洛隨機抽樣,分析青稞核心種質與等樣本隨機群體的多樣性差異。采用R程序t.test函數計算五個農藝性狀(穗長、芒長、株高、穗粒數和千粒重)在核心種質和1 220份材料間的差異顯著性。
2 結果與分析
2.1 群體結構和遺傳分化
通過MCMC模擬,當亞群數K逐漸增加時,貝葉斯模型輸出的后驗概率[LnP(D)]值呈逐步增加趨勢(圖1),并且LnP(D)值最明顯的變化發生在K由1 到2的過程中。分析貝葉斯后驗概率的變化速率(△K)發現,△K在K為2時出現峰值(圖 2)。綜合兩種方法,1 220份青稞可被劃分為兩個主要的亞群A1和A2(圖3)。A1亞群包含192個青稞品系,其中,29個品系來自歐洲和中亞地區,15個品系來自加拿大,13個來自墨西哥,9個來自澳大利亞,其余126個品系約有117個來自西藏地區。A2亞群包含1 028個青稞品系,其中,938個來自西藏地區,52個來自青海(29)和四川(23)地區,3個來自甘肅(2)、貴州(1),約9個來自加拿大和墨西哥等地。
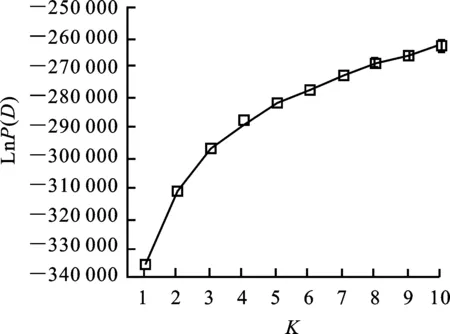
圖1 群體結構分析中貝葉斯模型后驗概率LnP(D)分布圖Fig.1 Posterior probability of Bayesian model [LnP(D)] in population structure analysis
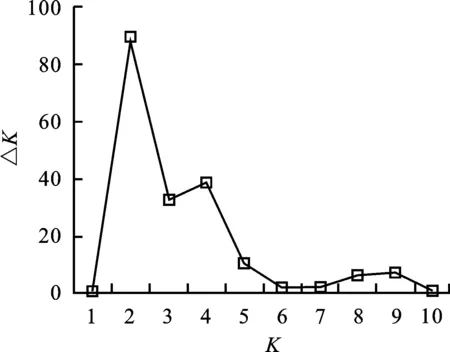
圖2 貝葉斯模型后驗概率變化速率(△K)分布圖Fig.2 Changing rate of posterior probability(△K)
△K在K為4時出現了第二個峰值(圖2),1 220份 青稞可進一步被劃分為4個亞群B1~B4。B4亞群包含194個材料,包括A1亞群的192個材料以及2個加拿大材料。A2亞群則被進一步被劃分為3個小亞群B1、B2、B3(圖3)。通過主成分分析(PCA),第一和第二主成分(principal component)分別能解釋1 220份青稞種質6.64%和4.96%的遺傳變異。A1和A2亞群的青稞材料具有較明顯的遺傳分化,而B1~B3亞群間的遺傳距離相對較近(圖4)。主成分分析結果與群體材料來源具有較好的吻合度。
利用AMOVA和pairwiseFST分析青稞不同亞群間的遺傳分化發現,不同亞群間遺傳分化程度差異顯著(P<0.001),A1和A2亞群間的遺傳分化能解釋17.39%的群體方差,而B1~B4亞群間的遺傳分化能解釋14.99%的群體方差(表1)。A1和A2亞群的分化系數FST為0.174(表2),表明我國和國外青稞種質因地理隔離和適應性等原因,具有明顯的遺傳差異。不同B亞群間的遺傳分化程度各不相同,其中,B4與B1、B2、B3亞群的分化系數較大(FST=0.191~0.198),而B1、B2、B3亞群間的分化系數較小(FST=0.105~0.131,表2)。這表明分布于我國不同地區的青稞種質間存在一定程度的遺傳差異,由于生態環境和育種需求等因素,其差異相對較小。
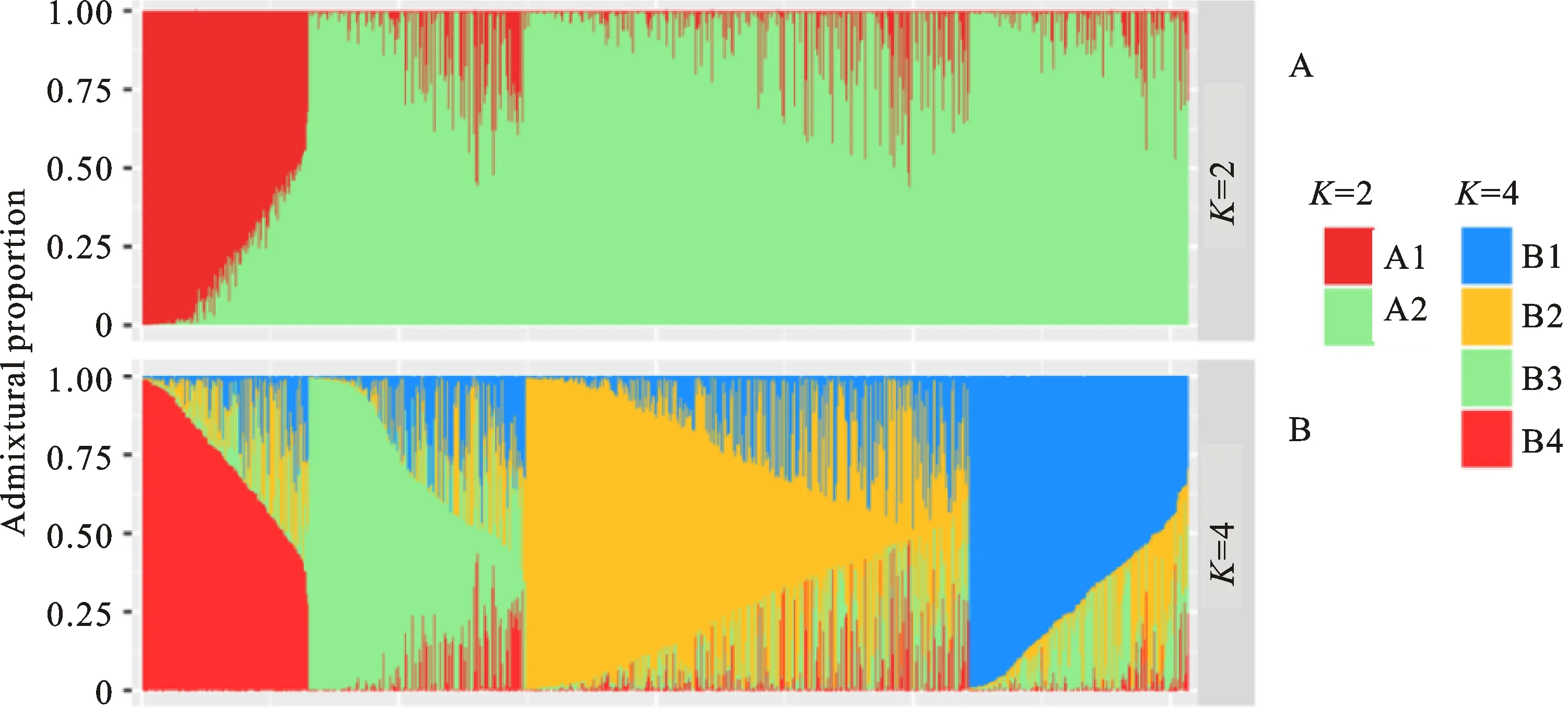
圖3 1 220份青稞種質資源群體結構分析。Fig.3 Admixture Proportion of 1 220 hulless barley accessions
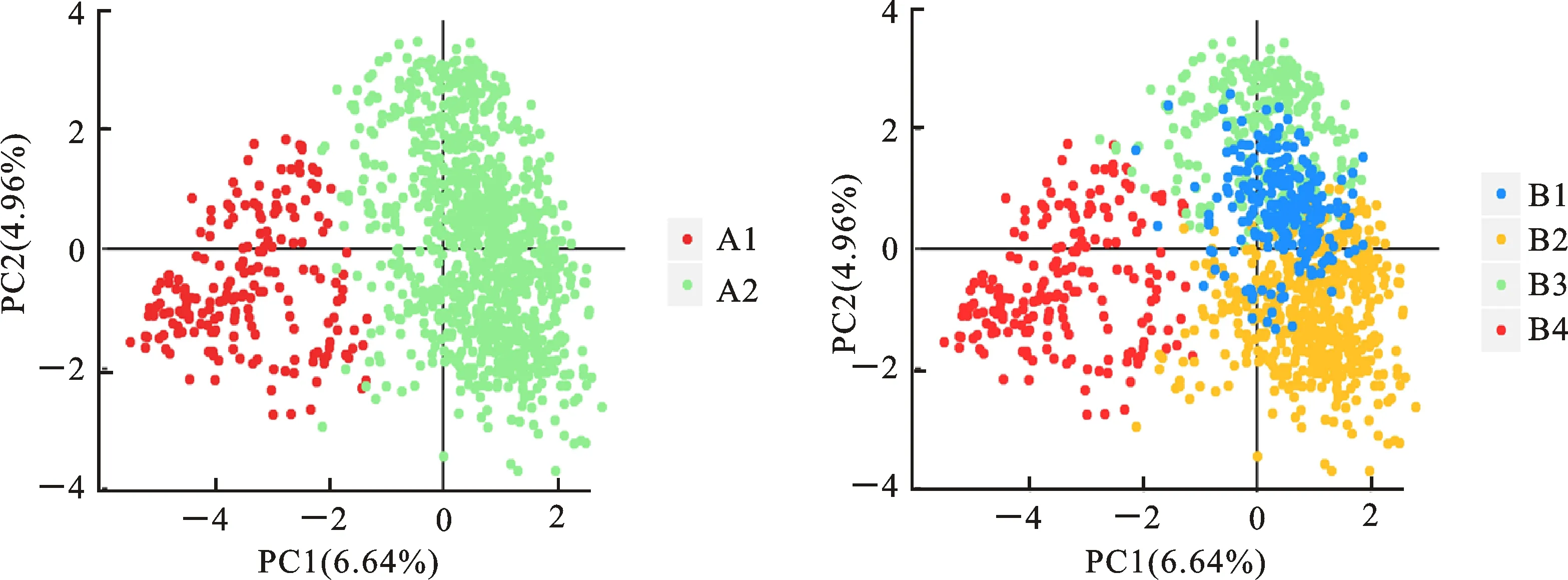
圖4 青稞群體主成分分析Fig.4 Principal component analysis of hulless barley germplasm
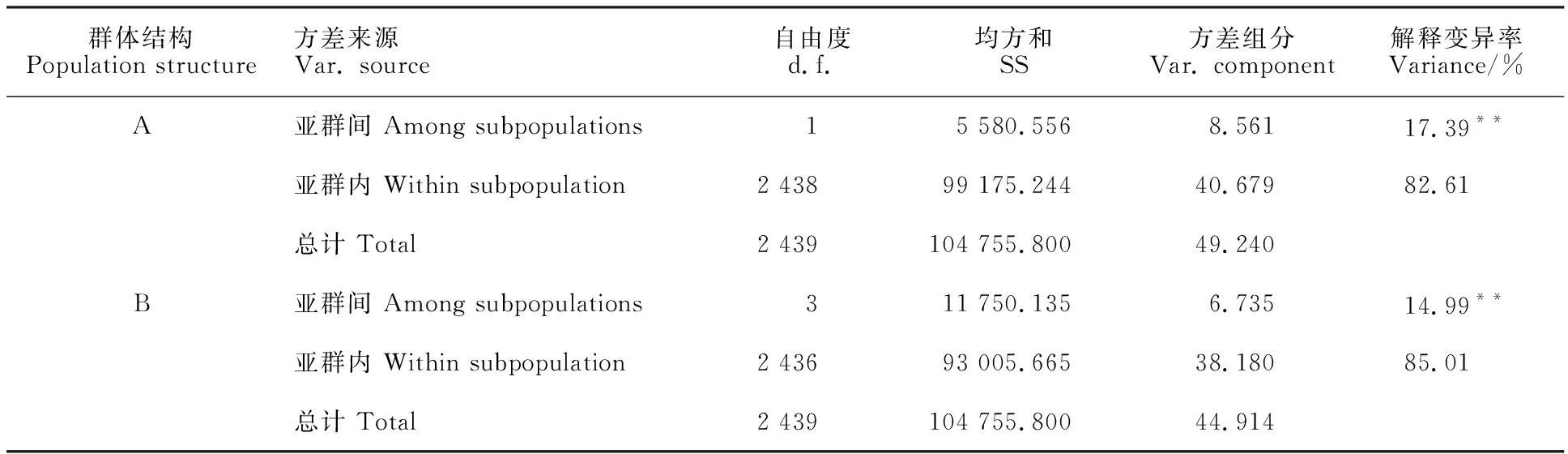
表1 青稞群體分子方差分析Table 1 Analysis of molecular variance(AMOVA) among subpopulations in hulless barley
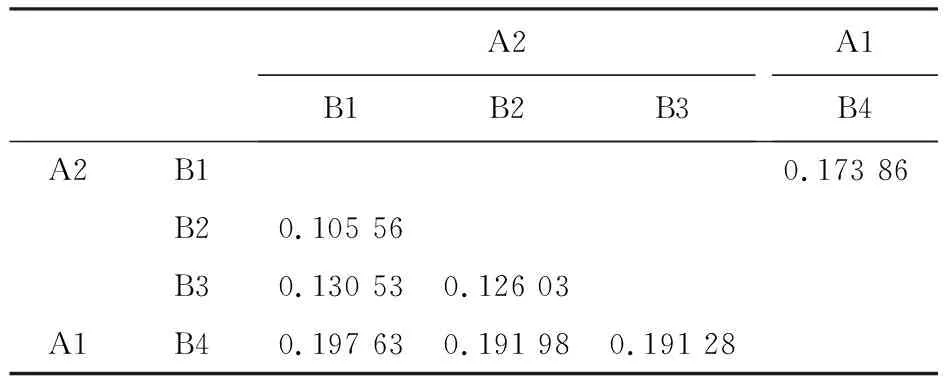
表2 不同青稞亞群間的分化系數Table 2 Genetic differentiation(FST) between hulless barley subpopulations
2.2 遺傳多樣性
95對SSR引物共檢測到451個等位變異,每引物平均檢測到4.74個等位變異,變化范圍為1~19。基因多樣性平均為0.22,變化范圍為0.02~0.50。多態性信息含量平均為0.18,變化范圍為0.04~0.80。等位變異豐度平均為0.37(表3)。A1亞群共檢測到418個等位變異,每對引物平均檢測到4.40個等位基因;而A2亞群共檢測到423個等位變異,每對引物平均檢測到4.45個等位變異。A1和A2群體間的SSR等位變異差異不顯著;其基因多樣性、多態性信息含量、香農系數和最小等位基因頻率均差異顯著。A2亞群的群體數約為A1亞群的5倍,A1亞群的變異豐度約是A2亞群的5倍。這表明A2亞群不同材料間的等位變異冗余度較高,具有進一步篩選有代表性材料的空間。在B1~B4亞群中,B4亞群多樣性顯著高于其他亞群。B2亞群包括517個青稞材料,共檢測到402個等位變異,等位變異豐度為0.78,約為B1和B2亞群的二分之一(表 3)。該結果表明,A2亞群中青稞材料的高冗余度主要由B2亞群材料導致。
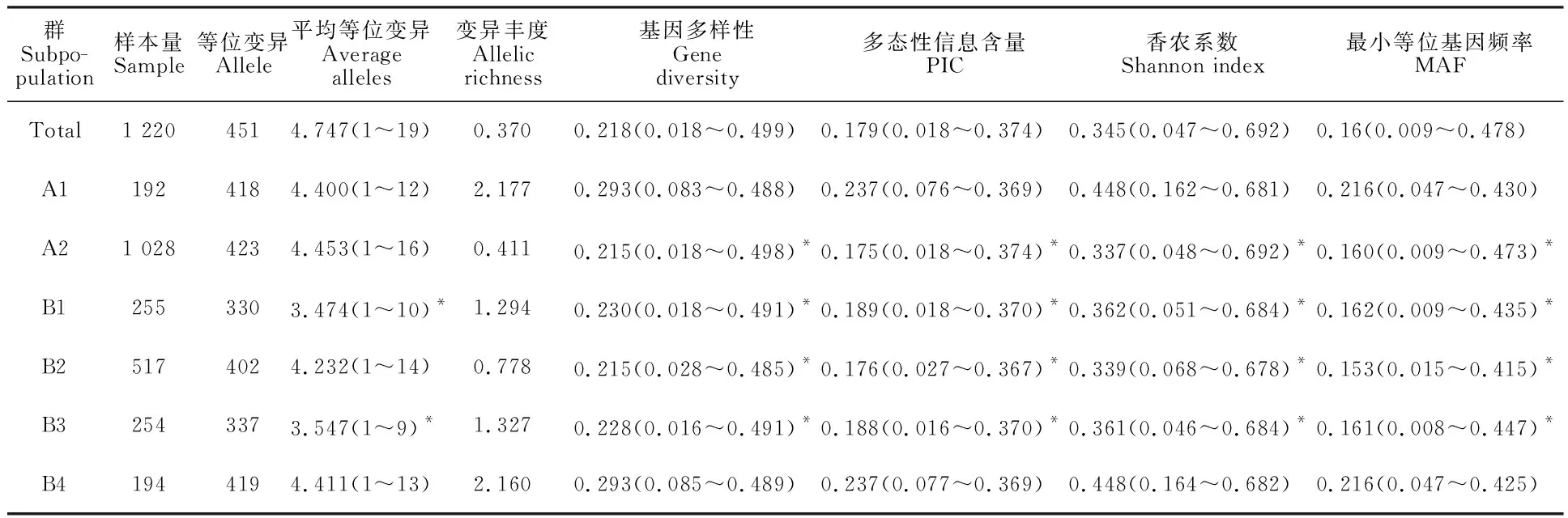
表3 青稞不同亞群遺傳多樣性Table 3 Diversity statistics of subpopulations in hulless barley
2.3 核心種質構建
分析樣本間遺傳距離和基因多態性,從1 220個青稞材料中篩選并構建出一個青稞核心種質群體,該群體包含300份材料,其中,163個材料來自A1亞群,占A1亞群材料數的84.9%;137個材料來自A2亞群,占A2亞群材料數的13.3%。
通過蒙特卡洛模擬實驗發現,從1 220個青稞材料中隨機抽取300個材料能攜帶平均415個等位變異,變異范圍為400~432,而青稞核心種質共攜帶444個等位變異,二者差異極顯著(P<0.001)。青稞核心種質的基因型多樣性指數平均為0.235,極顯著高于隨機群體(P<0.001)。青稞核心種質材料間的歐氏距離平均約為11.4,比原始群體間的歐式距離(8.2)明顯升高(圖5a)。說明該青稞核心種質能有效地代表1 220份原始材料。結合群體結構來看,在A1和B4亞群上,核心種質的遺傳多樣性與原始群體沒有明顯差別,核心種質的多樣性提升主要來自于A2亞群的變化(圖5b)。與此同時,最小等位變異頻率在核心種質中也同樣存在大幅提升,這也是群體多樣性增加的一個重要原因。
青稞核心種質的穗長、芒長、株高、穗粒數和千粒重均值與原始群體并無顯著差別(表4,P>0.05)。這表明青稞核心種質構建并未影響原始群體的表型分布。青稞核心種質的芒長和株高的標準差為2.89和21.49,分別為原始群體的95.7%和94.8%,而穗長、穗粒數和千粒重的標準差為1.82、18.62和6.03,為原始群體的1.08~1.18倍。該研究結果表明,青稞核心種質保留了原始群體絕大多數的表型變異,并因有效去除冗余材料,更有利于育種工作進行。
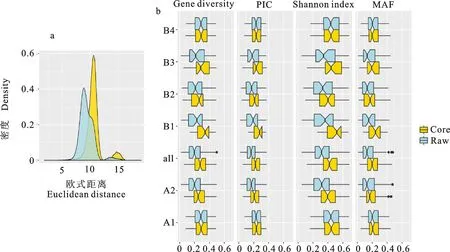
a:核心種質和原始群體的歐式距離分布圖;b:核心種質和原始群體不同多樣性參數的比較a:The distribution of Euclidean distances for core set and raw set;b:Comparison of genetic diversity between core set and raw set.
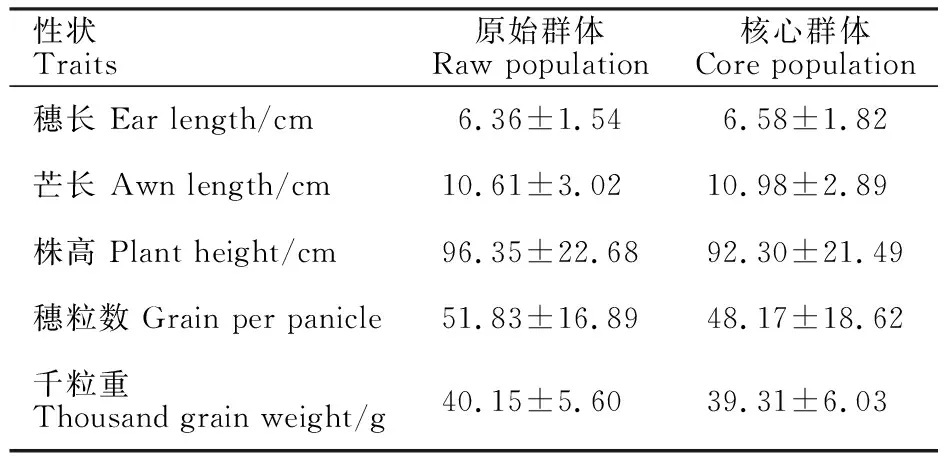
表4 青稞核心種質與原始群體的農藝性狀比較Table 4 Comparison of agronomic traits between hulless barley core set and raw population
3 討 論
國內外有眾多研究者對不同來源的青稞種質的遺傳多樣性進行過研究。楊 菁等[24]利用7個SSR引物對青海省42份栽培青稞進行遺傳了多樣性分析,每個SSR引物平均檢測到3個等位變異。范志芬等[25]利用30個SSR引物對64份青藏高原栽培青稞種質進行分析,發現栽培青稞有豐富的遺傳多樣性,平均每個SSR引物檢測到4.4個等位變異。楊 平等[26]對四川高原的25份青稞育成品種進行SRAP分子標記分析,每個SRAP平均檢測到5.2個等位變異,表明四川高原的青稞具有較高的遺傳多樣性。Pandey等[4]利用42個SSR引物對107個尼泊爾青稞材料進行基因型分析,發現尼泊爾青稞種質也具有較高的遺傳多樣性。
本研究中,1 220份青稞材料共檢測到451個等位變異,每個SSR引物平均檢測到4.7個等位變異。該結果比SRAP多態性水平略低,這可能是不同分子標記的擴增效率差異導致的。不同研究差異的可能原因如下:1)不同研究所用SSR引物的差異;2)擴增與檢測手段存在差異;3)研究群體遺傳背景的差異。隨著青稞參考基因組的公布和高通量基因型分析技術的發展,大麥已經開發出基于Illumina平臺的9K iSelect SNP芯片。這預示著高通量SNP分析技術的發展,對青稞的基因型鑒定、遺傳多樣性分析和重要性狀關聯分析具有重要的意義。
在農作物育種中,核心種質的篩選對雜交親本組配和大規模種子庫保種具有重要意義。核心種質能在一個較小的群體中最大化保留該群體的遺傳多樣性,從而提高育種效率。本研究從廣泛收集的1 220份青稞種質資源中,通過評估其遺傳多樣性和群體結構,構建了一個青稞核心種質群體。該青稞核心種質群體保留了原始群體98%的遺傳變異信息,極大地降低了材料間的冗余度,顯著提高了群體多樣性水平。除此之外,本青稞核心種質顯著地提升了群體最小等位基因頻率(MAF),這為后續基于核心種質的GWAS研究以及分子育種奠定了堅實的基礎。
