利用Illumina MiSeq高通量測序技術分析原料乳的菌群分布
2018-08-31 02:32:30于國萍姚宇秀范美婧董良偉
食品科學 2018年16期
于國萍,陳 媛,姚宇秀,范美婧,劉 鵬,董良偉
原料乳中營養成分比較高,是各種細菌滋生的絕佳環境,原料乳可以為其提供充足的養分和合適的pH值,所以原料乳中有著相對密集的微生物[1],其數量從幾百到幾千CFU/g不等[2]。其中部分微生物能夠改善乳制品的口感等,這些微生物并不都是有益的,有的是有害的,可以引發比較嚴重的疾病[3]。原料乳中常見的微生物有3 大類,即有益微生物、有害微生物和病原微生物[4]。
原料乳中有益微生物主要是乳酸菌,而乳酸菌主要包括鏈球菌、明串珠菌和乳桿菌屬等。有害微生物常稱為腐敗菌,其中最主要的就是嗜冷菌。病原微生物通常是致病菌,它即便不改變乳的性質,卻也可以造成疾病,引發大規模的感染。原料乳中食源性致病菌主要為大腸桿菌O157:H7、沙門菌、單核細胞性李斯特菌、金黃色葡萄球菌以及志賀菌等[5-8]。
由于乳及乳制品具有較高的營養成分,我國對其的需求越來越大,因此對原料乳的微生物的檢測也比較重要[9],原料乳中微生物多樣性的檢測方法也從原來的純培養手段轉變為分子生物學手段[10-11]。Fricker等[12]采用純培養和分子方法相結合的手段研究了微生物的多樣性,Bonizzi等[13]采用了內轉錄間隔區序列分析了微生物的構成,Raats等[14]采用變性梯度凝膠電泳(denaturing gradient gel electrophoresis,DGGE)和克隆文庫的方法對原料乳及其冷藏過程中菌群的變化進行研究。其中,純培養的手段比較繁瑣,而DGGE等分子生物學技術很難全面地反映微生物的多樣性。
隨著乳及乳制品的大量生產,我國乳制品的質量問題經常出現[15],這很大程度上是由于乳及乳制品中感染了致病菌,導致食源性疾病的發生。因此,加強原料乳的質量安全檢測至關重要,可以防止大規模食源性疾病的發生[16-17]。目前使用的檢測方法大多為分子方法[18-22],已有研究分別采用不同的聚合酶鏈式反應(polymerase chain reaction,PCR)技術檢測了乳中致病菌。雖然目前的分子檢測方法有很多優勢,但是仍舊檢測不全面,因此本實驗采用高通量測序(high-throughput sequencing,HTS)手段對原料乳的菌種進行檢測。
HTS又叫“下一代”測序技術[23-24],主要是能同時對很多樣本中提取的DNA進行測序,效率非常高。HTS技術給過去分子水平的生物測序技術帶去了革命性的創新,已經普遍利用到關于基因組測序等一系列分子水平的測序方法上[25-27]。本實驗主要采用的是Illumina公司的MiSeq測序平臺[28],MiSeq個人測序系統采用Illumina的TruSeq邊合成邊測序技術。第2代測序Illumina MiSeq方法分析有效的避免了通量低、操作復雜和準確率低等缺陷[29-32],具有操作簡單、成本較低的優勢,并且采用邊合成邊測序原理,結果可信度高。
本實驗主要是利用HTS的方法測定原料乳中菌種的分布情況。采集14 個奶牛場的原料乳,通過HTS的方法(采用Illumina MiSeq系統)測定菌群多樣性分布情況,然后根據測序結果,了解原料乳中菌群的分布情況,為構建奶牛養殖風險防控機制,并提出防控奶牛養殖風險的對策提供參考。
1 材料與方法
1.1 材料與試劑
原料乳 黑龍江省內14 個奶牛場提供。
E.Z.N.A.Soil DNA試劑盒 瑞士Omega公司;Qubit 2.0 DNA檢測試劑盒 美國Life公司;Taq DNA Polymerase 美國Thermo公司;Agencourt AMPure XP美國Beckman公司。
1.2 儀器與設備
Pico-21型臺式離心機 美國Thermo Fisher公司;GL-88B型旋渦混合器 海門市其林貝爾儀器制造有限公司;TND03-H-H型混勻型干式恒溫器 深圳拓能達科技有限公司;DYY-6C型電泳儀電源、DYCZ-21型電泳槽北京市六一儀器廠;凝膠成像系統 美國UVP公司;Q32866型Qubit?2.0熒光計 美國Invitrogen公司;T100TMThermal Cyeler型PCR儀 美國Bio-Rad公司。
1.3 方法
1.3.1 樣品的采集
在黑龍江省(包括哈爾濱、雙城、阿城等地)14 個奶牛場采集原料乳,將樣本放在-20 ℃的冰箱里面凍存,并進行后續的實驗,按采集的奶牛場的順序分別標記為1~14。
1.3.2 樣品的前處理
將凍存后的樣品進行解凍,因為菌株樣品和原料乳屬于液體,所以其中包括的菌種的數目相對較低,進行后續實驗之前一定要將樣本混合均勻,樣本的菌種比較豐富時,吸取一定量的樣本進行離心,倒出液體,留下離心后的菌種。
1.3.3 DNA的提取
DNA的具體提取步驟參照E.Z.N.ATM Soil DNA試劑盒進行,按說明書操作。所提取的DNA于-20 ℃保存備用。
1.3.4 瓊脂糖電泳
將提取的DNA進行瓊脂糖電泳。將一定量的DNA與一定量的上樣緩沖液按比例混合,然后吸取其中的混合液10 μL進行DNA電泳,條件為200 V、45min,在凝膠成像系統中進行觀察。
1.3.5 PCR擴增
1.3.5.1 第1輪擴增
PCR采用雙引物進行擴增,引物序列是V3-V4通用雙引物[33]。主要是先將DNA產物加入試劑盒中的各項物質形成1 個體系,需要加入的物質為PCR緩沖液、核苷酸、DNA樣品、Bar-PCR primer F、Primer R、Plantium Taq、雙蒸水,使整個反應體系體積為50 μL。
將混合好的物質放入PCR儀中進行擴增,主要步驟是:先進行預變性,然后進行變性過程,需要將其循環多次,最后延伸即可。之后進行第2輪擴增。
1.3.5.2 第2輪擴增
引入Illumina橋式PCR兼容引物,第2次PCR需要加入的物質與第1次類似,只是加入的量不同,同樣是使整個反應體系是50 μL。擴增的步驟與方法見1.3.5.1節。
1.3.6 PCR產物的瓊脂糖電泳
將PCR后物質進行DNA電泳,步驟同1.3.4節。
1.3.7 DNA純化回收
將擴增后物質進行DNA的純化回收,不同微生物的擴增后的物質采用的回收的方法基本一致,不同的是選擇的磁珠不同。具體步驟是:用磁珠對擴增后的產物進行吸附,吸附3 次,在吸附的過程中,需要將其放在磁力架上,保證它的吸附力。最后用洗脫液洗脫后保存到小的EP管中,進行后續的實驗。
1.3.8 定量混合
利用Qubit 2.0 DNA檢測試劑盒對回收的DNA進行定量。最后要將純化后的DNA與原來的進行混合,使最后進行測序的物質濃度為20 pmol/L。
1.3.9 數據預處理
1.3.9.1 原始序列數據
經過系統整理的數據可以經過一個系統進行轉化,成為最初的測序序列,即Raw Data,將其保存到電腦中,它是由所有的序列信息和與其相關的質量數據組成。
1.3.9.2 數據預處理
經過HTS后的數據含有多余的序列,所以在處理數據之前應該先將多余的序列除去,然后才可以進入系統中進行測序。開始先將多余的引物除去,之后成對的read拼接成一條序列,將序列通過系統識別,將不同樣本的數據分開,最終對樣本中的數據進行檢查,去掉不合格的數據,留下最后的有效數據。
1.3.10 去除嵌合體及非特異性擴增序列
在進行PCR擴增的時候,有時候會出現嵌合體,它通常是由于在第1輪擴增過程中的延伸階段,并沒有完全進行,只反應到了一半,使得產物不完整,這些不完整的產物會進行下一次擴增,可能會使其加入一些與之不同的片段,形成一種雜交的DNA,這就是嵌合體。同時也會產生一些非特異性擴增序列。為了確保結果的準確性,需要將這些嵌合體除掉。使用Usearch去非擴增區域序列。先采用uchime驗證這個序列是嵌合體,驗證成功后將其去除。
1.3.11 分析項目
操作分類單元(operational taxonomic units,OTU)聚類分析:依據每個序列之間的遠近程度進行分析,將距離近的歸為一個OTU。選擇其中最典型的序列進行檢測,通常選擇序列數目多的作為典型性序列,然后根據OTU的分類情況進行了聚類分析。
Alpha多樣性分析:衡量樣本物種多樣性。計算物種并制作所有樣品豐富度指數、Chao、Shannon、Simpson、Coverage等物種多樣性指數,箱形圖和稀釋性曲線。根據各樣本的OTU豐度分布情況繪制相對豐度曲線。
物種分類分析:將樣本中的序列進行分類后,分類后的序列屬于一個菌屬,然后根據菌屬的數量進行分析。基于物種分類分析,繪制物種分類條形圖、物種豐度餅圖、物種豐度熱圖、單樣本群落分布豐度柱狀圖、群落分布分度3D圖、樣本聚類與柱狀圖組合分析圖、菌群分布條形圖等。
1.4 數據分析
利用Mothur 1.30.1軟件、FLASH 1.2.3軟件、pear 0.9.6軟件、Usearch 5.2.236軟件、Cytoscape 3.2軟件、R 3.2軟件等進行數據處理分析。
2 結果與分析
2.1 OTU基本情況
將提取的DNA進行電泳,并將擴增后的產物進行電泳,目的是檢測DNA的完整性及其是否能進行后續的實驗,擴增后的條帶比較亮,說明可以進行后續的實驗。OTU指在生物學系統中,為了詳細認識樣本中菌種的具體分布情況,要將測序的結果按種類開始統計,人為規定一個分組的單位稱為一個OTU。一般情況下,認定2 個序列相似度為97%時屬于一個菌屬,當相似度為99%時,認定其是一個菌種。本實驗采用的相似度為97%為一個OTU。
2.2 基于OTU豐度的樣本聚類圖
樣本聚類樹圖可以直觀地反映樣本間的相似度,具體方法如下:首先使用描述群落組成關系和結構的算法計算樣品間的距離,依據常用的一種算法創造一個或者多個節點的圖譜,算法是非加權組平均法,算出一個結構圖譜,可以直接觀察圖譜分析數據,計算樣本間距離的方法為布雷柯蒂斯距離。在目前的統計學里,聚類分析全是利用布雷柯蒂斯距離分析的,它能夠顯示菌落差異,因此本實驗使用該參數反映菌落的差異。原料乳的14 個樣本的基于OTU的樣本聚類樹圖見圖1,樹枝的長度代表樣本間距離,分支的寬度表示樣本間的距離的遠近,相似度高的樣本距離越小。由圖1可以看出,原料乳的14 個樣本類聚成2 個主要群體,3、4、5、6、7、8、9、10、11是一類,1、2、12、13、14是另一類。其中12和13是最相似的兩個樣本。整體看14 個樣本,相似度差別略大,可能是由于在黑龍江省不同的區域進行取樣,地域的差異性導致的。

圖1 基于OTU的樣本聚類樹圖Fig. 1 Clustering dendrogram based on OTU samples
2.3 稀釋性曲線
為確定所得到的結果正確性,是否能進行后面的實驗,需要進行稀釋性曲線分析,它主要是從整個樣品的數據里面抽取部分進行分析,分析其所包含的菌種的多少,是通過抽取的個體與物種的數目建立圖譜。通過該曲線能夠分析樣品的菌種數是否豐富,也可以分析結果是否正確。隨機選擇樣本中的一部分序列,將其指代的OTU與序列數繪制稀釋曲線,如圖2所示,沿著橫坐標曲線逐漸穩定時,看出結果分析比較合理,相反,如果曲線不穩定,說明結果不合理,不能進行分析。所以,通過觀察該曲線,能看出樣本是否檢測完全。整個曲線都是依據相似度97%繪制的。由圖2可以看出,所有樣本的曲線都隨著樣本中隨機抽取序列數增大而逐漸穩定,說明結果比較合理,所含菌種數較多,可以進行后續的測序。
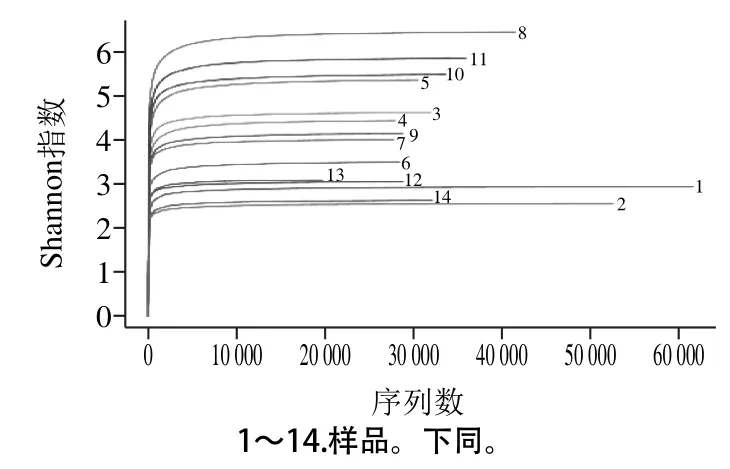
圖2 Shannon指數稀釋分析圖Fig. 2 Rarefaction curves for Shannon index
2.4 Rank-abundance曲線
為了直觀地表示每個樣本中菌種的數目是否豐富,進一步繪制了Rank-abundance曲線,它與多樣性指數一樣,可以看出樣本的多樣性。主要是計算每個樣本中的各個OTU包括的序列數目,按照從大到小進行排列,繪制曲線如圖3所示。當然,Rank-abundance曲線能夠表示樣本菌種數目的兩大部分,包括樣本中所含的菌種的豐度及其是否均勻。曲線的寬度代表了菌種的豐度,曲線越寬,說明所包含的菌種越多;曲線的平穩程度代表了其菌種數目是否均勻,曲線越穩定,說明菌種的構成越均勻。由圖3可以看出,所有的樣本在橫坐標上的寬度的范圍分布較廣,說明所含菌種的數目較多,組成豐富;同時可以看出曲線比較穩定,說明菌種的構成比較均勻。
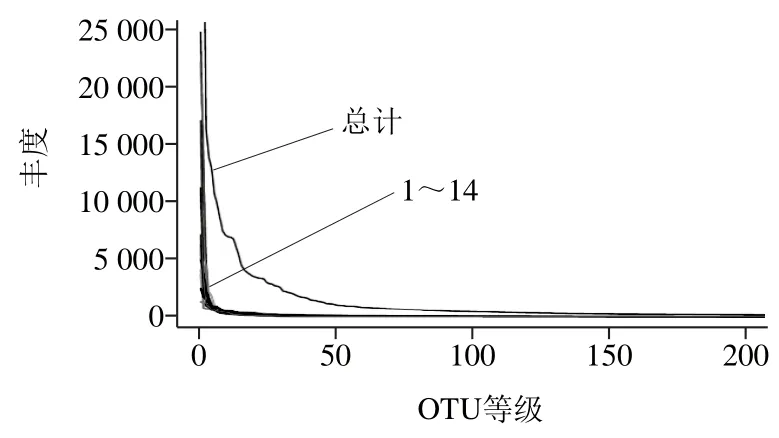
圖3 Rank-abundance曲線圖Fig. 3 Rank-abundance curves
2.5 物種累積曲線
為了確定樣品中的菌種的數目是否會根據樣本的數目增加而增加,繪制了物種累積曲線圖,由其可看出測序的菌種數是否完全檢測出來,也可以看出樣本中的菌種數量是否滿足需要。根據曲線的走向不但能夠知道樣本中菌種是否足夠,也可以看出樣品中物種的豐度。物種累積曲線代表了隨著樣本的增加是否會有新的OTU出現。在曲線中,隨著樣本的增加,如果曲線快速增長,說明有更多的OTU檢測出來;如果曲線增長緩慢,說明沒有更多的OTU檢測出來,說明結果合理。因此通過該曲線能夠判斷樣品量是否滿足需要,如果曲線快速增長,需要加入足夠的樣品量滿足測序分析,相反的話,就可以直接進行后續的實驗。由圖4可以看出,沿著橫坐標樣本的增多,曲線增長比較緩慢,說明沒有更多的OTU檢測出來,說明樣品量足夠,可以滿足需要。
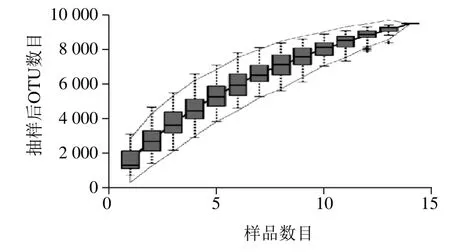
圖4 物種累積曲線圖Fig. 4 Species accumulation curves
2.6 物種豐度熱圖
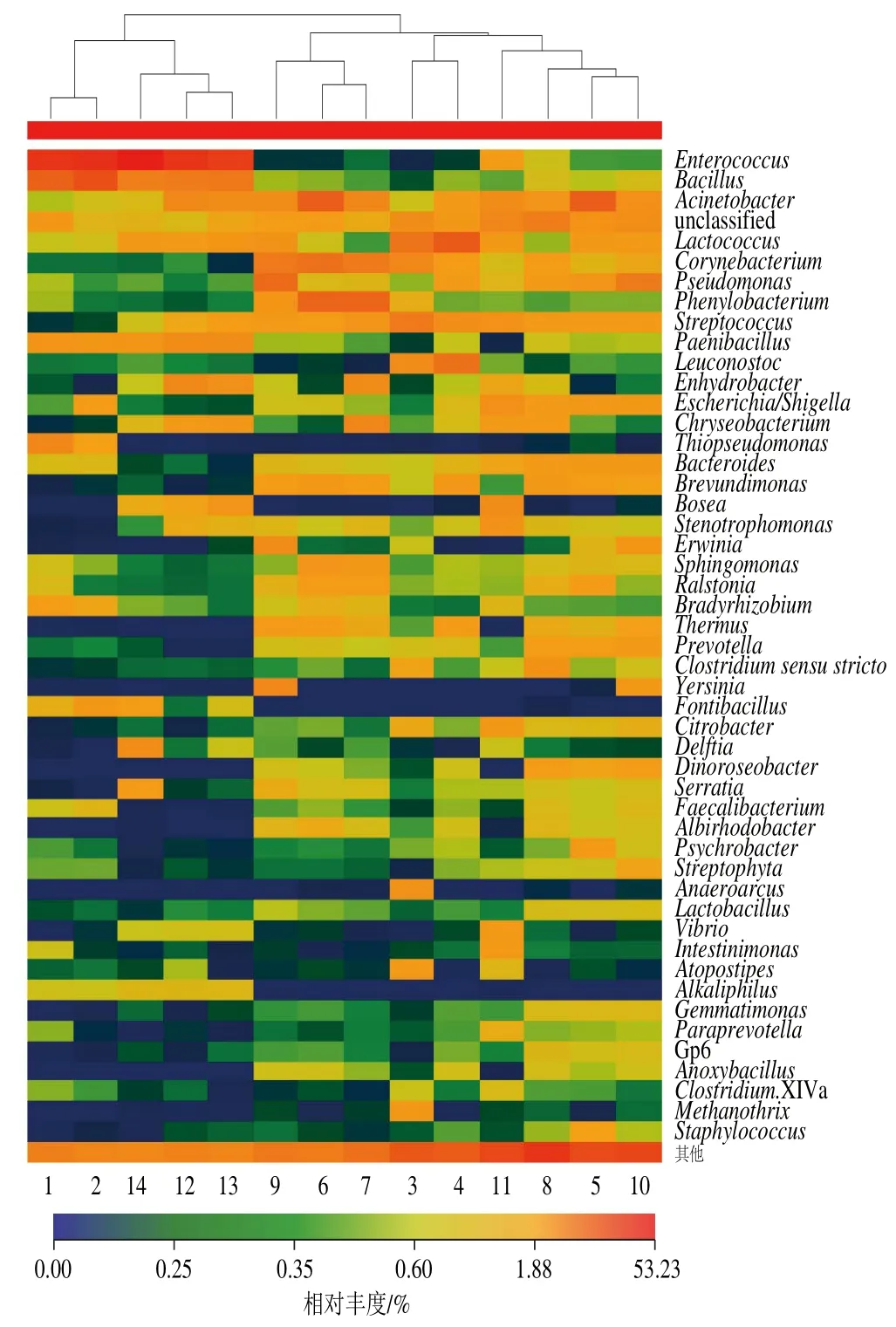
圖5 屬水平物種豐度熱圖Fig. 5 Heat map of species abundance at genus level
菌群數量用熱圖顏色標記,曲線制作過程是將菌落的數量聚類分析,把分析后的結果體現在熱圖中。原料乳中14 個樣本的屬水平物種豐度熱圖,如圖5所示,顏色越紅說明所含的該菌種的數量越多,顏色越藍則說明所含的菌種不多。為了展示效果,只顯示豐度最高的前50 個物種分類信息,剩余的物種分類合并成其他。由圖5可以看出,14 個樣本的前5 種菌種的顯示顏色比較紅,含量比較多,分別是腸球菌屬、芽孢桿菌屬、不動桿菌屬、乳球菌屬,其中第4個豐度較高的菌屬并沒有與之匹配的數據庫。其他菌屬中有的樣本顏色比較紅,有的樣本顏色比較暗,說明每個樣本中菌群的豐度不同,可以看出所有樣本的菌群的多樣性。
2.7 所有樣本菌群分布條形分析

圖6 原料乳的菌群分析條形圖Fig. 6 Barplot for bacterial genus distribution in raw milk
依據微生物的特性,重點分析菌種的菌群分布,通過菌群分布條形圖可以看出菌種的大概分布,所占的比例等,另外還可以看出不同的樣本之前的差異。由于菌群的結構圖包括單樣本群落分布豐度柱狀圖,群落分布分度3D圖,樣本聚類與柱狀圖組合分析圖、菌群分布條形圖等,本實驗用菌群分布條形圖來代表。由圖6可以看出,原料乳的14 個樣品的菌種的分布情況。1、2、12、13、14號奶牛場的原料乳中腸球菌屬、芽孢桿菌屬較多,3、4號奶牛場中乳球菌屬較多,5號奶牛場中不動桿菌屬較多,6、7號奶牛場中苯基桿菌較多,8號奶牛場中梭狀芽孢桿菌較多,9、10號奶牛場中假單胞菌屬較多,11號奶牛場中慢生根瘤菌科中的一個屬較多。在結果中關于致病菌的菌種和菌屬,可以得知1、2中有金黃色葡萄球菌,2、5、8、10中有志賀菌,其他樣本的致病菌暫時沒有檢測出來。
3 結 論
本實驗率先將HTS技術應用在原料乳的菌群的研究中,通過HTS技術了解原料乳中菌群的分布情況。由此可知不同牧場原料乳的菌群存在多樣性,位于前列的分別是腸球菌屬、芽孢桿菌屬、不動桿菌屬、乳球菌屬,部分原料乳中有金黃色葡萄球菌和志賀菌的存在。相關領域研究者采用不同的方法對原料乳菌群分布進行研究,例如,李博等[34]利用宏觀菌落形態學觀察的方法檢測原料乳中的主要微生物,即在鑒別培養基上培養菌群,觀察生長情況,結果表明原料乳中含有乳酸菌、大腸桿菌、嗜冷菌;Pourhanssan等[35]采用革蘭氏染色結合鏡檢的方法對原料乳中的菌群進行了分析鑒定,結果表明原料乳中有大腸桿菌、金黃色葡萄球菌、腸桿菌、克雷白氏桿菌屬、綠膿桿菌以及變形桿菌屬;相較于HTS,分離培養、生化鑒定無法對難培養或者不可培養的致病菌進行檢測,存在特異性差、操作復雜、耗時等缺點,無法實現及時有效檢測。劉洋等[36]利用基質輔助激光解吸電離飛行時間質譜鑒定原料乳中細菌菌落分布,鑒定出原料乳中含有乳酸菌、腸桿菌、金黃色葡萄球菌;與這些方法相比,HTS的研究結果更加全面地反映了原料乳菌群分布多樣性及豐度信息。此外,夏圍圍等[37]利用HTS和DGGE分析土壤微生物群落結構的比較研究中,得出HTS能夠較為全面和準確地反映土壤微生物群落結構,而DGGE僅能分析有限的優勢微生物類群,存在高估物種豐度以及低估微生物群落大小和多樣性的可能。因此應該嚴格控制奶牛生活環境的衛生情況,防止金黃色葡萄球菌和志賀菌污染原料乳,保障原料乳質量安全,為有關部門控制原料乳及儲奶設施的衛生條件提供有力的理論和實驗依據。
本研究利用HTS方法,對原料乳的菌群分布情況進行了較全面、系統、準確的檢測,具有很高的特異性和靈敏度,并為原料乳中致病菌檢測提供了技術支持。隨著HTS技術的不斷推進,相信在不久的將來,它會在食品的檢測領域有更廣泛和全面的應用前景。
猜你喜歡
英語世界(2023年10期)2023-11-17 09:18:18
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
電子制作(2018年18期)2018-11-14 01:48:24
汽車觀察(2018年10期)2018-11-06 07:05:26
山東工業技術(2016年15期)2016-12-01 05:31:22
海峽科技與產業(2016年3期)2016-05-17 04:32:12
