基于色譜-質譜法的肉類食品中大環內酯類藥物殘留及其代謝產物分析方法研究進展
2018-09-06 10:53:56許秀麗儲曉剛
分析測試學報 2018年8期
關鍵詞:檢測
賈 瑋,王 菡,石 琳,許秀麗,儲曉剛,
(1.陜西科技大學 食品與生物工程學院,陜西 西安 710021;2.中國檢驗檢疫科學研究院,北京 100123)
隨著許多食品安全事件的發生,食品安全問題日益受到關注,我國《“十三五”國家食品安全規劃》為進一步提升食品安全標準管理水平,深入開展了農獸藥殘留綜合治理工作,其中獸藥殘留問題是我國食品藥品監管部門重點控制的問題之一[1-2]。大環內酯類藥物(Macrolide drugs,MALs)是獸藥中非常重要的一類廣譜抗菌性抗生素,普遍應用于治療豬、羊、牛和家禽的呼吸性及傳染性疾病,或在低劑量下作為飼料添加劑促進動物生長發育[3]。但長期食用會導致大環內酯類藥物及其代謝物在動物組織及器官內蓄積或儲存,當該藥物在人體內累積達一定濃度時,可引起胃腸道不良反應和耳蝸神經損害,嚴重者還會造成肝腎的損傷[4-5],此外,食品中的大環內酯類抗生素殘留不僅易造成致敏和毒性反應,致使細菌耐藥性升高,引起攜帶耐藥因子菌株的擴散[6-7],殘留的藥物進入環境中還會導致嚴重的環境問題[8]。歐盟委員會[9]、我國農業部[10]、美國[11]、日本[12]等均對肉類食品中的大環內酯類藥物及其最大殘留限量(MRL)進行了限制(見表1),但各國監管機構均未發布針對大環內酯類藥物代謝產物的分析方法。開展動物源性食品中大環內酯類藥物及其代謝產物殘留的監測工作可為制定和完善國家標準提供參考,對于保障肉類食品的食用安全,保障人類健康及肉類食品產業發展具有重要意義。本文介紹了大環內酯類獸藥分析檢測的背景現狀,以及大環內酯類藥物進入畜禽類動物體內形成的代謝產物及其分布部位,對國內外肉類食品中大環內酯類獸藥殘留的前處理方法及檢測方法進行研究與探討,同時對大環內酯類藥物及其代謝產物殘留分析的研究方向進行了展望。

表1 各國及地區對大環內酯類藥物的最大殘留限量規定[9-12]Table 1 MRLs of macrolides in different countries and regions[9-12] w/(μg·kg-1)
EU:歐盟;CHN:中國;USA:美國;JPN:日本
1 大環內酯類藥物殘留檢測研究現狀
目前在Web of science數據庫中,以macrolide(and) detection(or) veterinary drugs(and) detection為主題共檢索出10 835篇文獻,近5年約占29%,如圖1A所示。在中國知網數據庫中,以大環內酯為檢索詞全文檢索,共檢索出10 640篇,近5年約占44%,如圖1B所示。總體上,近10年國內外對大環內酯類抗生素的研究均明顯增多,近5年國內外有關大環內酯類藥物研究的文獻比例均較高,表明大環內酯類藥物正引起人們的日益關注。
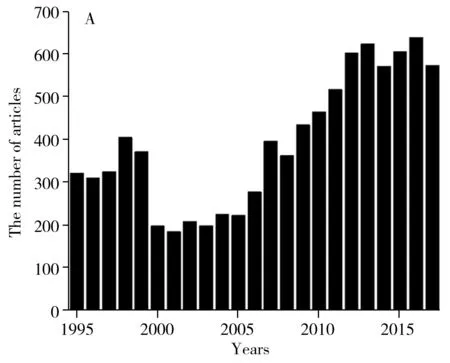
2 大環內酯類藥物代謝產物及其分布
獸藥殘留是指給動物使用獸藥后,蓄積或貯存在動物細胞、組織或器官內的藥物原形、代謝產物和藥物雜質[13]。大環內酯類藥物進入動物機體后,經腸胃的消化會產生復雜的代謝產物,代謝物保留了原有的活性基團,仍具有與原藥相似的抗菌活性,部分代謝產物的潛在危害甚至高于原藥形式。藥物原形和代謝產物通常以游離或結合的形式殘留在畜禽產品中[14]。目前國內外對大環內酯類藥物的研究多為原藥檢測,對其代謝物的檢測報道較少。為檢測食品中的大環內酯類獸藥殘留總量,需對大環內酯類藥物代謝物及其分布進行研究。常見的大環內酯類藥物有泰樂菌素、替米考星、西地霉素、紅霉素、竹桃霉素、螺旋霉素、吉他霉素、交沙霉素等。

西地霉素在機體內易發生代謝,代謝物有20多種,主要為蘭卡殺菌素C、蘭卡殺菌醇和蘭卡殺菌醇A。西地霉素原藥和代謝產物在動物機體內的滯留期短,畜類肝中的藥物殘留最高,肌肉和脂肪中未檢出殘留[18]。紅霉素廣泛分布于各種組織及體液中,并以在膽汁和肝臟中的含量最高,大部分藥物在肝臟代謝[19],主要代謝途徑為脫氧糖胺的N-去甲基化,代謝物主要為脫水紅霉素A和紅霉素A烯醇醚等[20]。竹桃霉素內服一般用三乙酰竹桃霉素,三乙酰竹桃霉素可在動物機體內代謝,而竹桃霉素在體內不代謝,在大部分組織如肝、腎中可檢測到殘留物,主要代謝產物為醋竹桃霉素。因此,檢測大環內酯類藥物的代謝物對監控此類藥物殘留更具實際意義。
螺旋霉素經機體吸收后廣泛分布于體內,主要代謝產物為新螺旋霉素。螺旋霉素于肝臟代謝,在動物的肝和腎中殘留最高[21]。吉他霉素的機體吸收良好,廣泛分布于主要的臟器,尤其以肝臟和腎臟中的濃度最高,主要經肝膽系統排泄,在膽汁和排泄物中的濃度高,少量經腎臟排泄。吉他霉素的代謝途徑為羥化,其在腸道吸收快,釋出也快,因此是在動物組織中殘留量最低的一種大環內酯類藥物。交沙霉素機體吸收迅速,殘留物在組織和臟器中濃度高,特別是在膽汁和肺中濃度較高,主要經膽汁排泄[22]。
目前為止,僅有少量文獻對同時檢測大環內酯類抗生素及其代謝物進行報道[23-25]。因此,有必要建立多殘留分析方法測定肉類食品中大環內酯類藥物及其代謝產物的殘留。
3 大環內酯類藥物及其代謝產物的分析方法
3.1 樣品前處理
為制備適用于色譜或液相色譜-質譜(LC-MS)法的供試液,需對樣品進行前處理。首先對樣品進行研磨或勻漿處理,然后進行提取、凈化、濃縮等步驟。由于肉類食品基質的復雜性,一部分大環內酯類藥物殘留與基質中的大分子(蛋白質等)結合,引起嚴重的基質干擾,故前處理必須分離藥物與蛋白質的結合體,以除去蛋白質等雜質的干擾,最大限度地提取大環內酯類藥物殘留。目前常見的前處理方法有液-液萃取(Liquid-liquid extraction,LLE)、固相萃取(Solid phase extraction,SPE)、QuEChERS(Quick、Easy、Cheap、Effective、Rugged、Safe)、分子印跡(Molecular imprinting,MI)、加壓溶劑萃取(Pressurised liquid extraction,PLE)、在線固相萃取(Online SPE)等。
3.1.1液-液萃取法液-液萃取通過加入一定的極性或非極性有機試劑進行提取,一般采用乙酸乙酯、氯仿等進行提取后過膜處理,除去雜質,用流動相溶解后進樣分析。岳振峰等[26]在聚丙烯離心管中用乙腈萃取動物組織中的大環內酯類抗生素,然后以正己烷脫脂,旋轉蒸發濃縮后進行測定,得到肉類食品中的回收率為60%~99%。液-液萃取法可用于大環內酯類藥物殘留的定量檢驗,對實驗條件和儀器要求不高,但該方法的凈化效果相對較差、操作費時、有機溶劑消耗大。
3.1.2固相萃取法固相萃取法是最常用的凈化方式,該方法是將待測物質從不同樣品中分離后,經過不同類型的固相萃取進行富集、凈化及除雜[27-28]。目前,已有多種SPE柱被用于大環內酯類藥物的純化,如C18柱[29]、HLB柱[30]、PEP柱[31]、Prime HLB柱[32]等,且均能有效地分離純化肉類食品中的大環內酯類藥物。目前常采用液相萃取結合固相萃取的方法進行大環內酯類藥物及代謝產物殘留的檢測,Cao等[33]在檢測家禽肌肉中獸藥殘留物的過程中,采用PSA和NH2去除提取物中的脂質和碳水化合物,乙腈-乙醇水溶液萃取,正己烷純化,定量下限為0.05~10 μg/kg。該方法符合歐盟委員會決議2 002/657/EC的標準并已成功用于當地市場(中國)的家禽肌肉樣品分析。
3.1.3QuEChERS法QuEChERS法因快速、高效、成本低等特點被廣泛用于農藥檢測領域,近年來也逐漸應用于獸藥殘留檢測,但在肉類食品的大環內酯類藥物殘留檢測中應用較少[34-35]。郭海霞等[36]通過一次提取分步凈化的方式,實現了121種獸藥的同時分析,該方法對C18、PSA、NH2和GCB 4種吸附劑的凈化效果進行了比較,發現C18和PSA結合的效果最好,可更好地去除基質中的有機酸、糖類等干擾物,極大地降低了分析費用,縮短了檢測周期,對肉類加工企業的質量安全控制具有較高的實用價值。
3.1.4分子印跡技術分子印跡技術在獸藥殘留分析領域是一種新興的分析手段。分子印跡聚合物(MIP)制備簡單,穩定性好,能重復使用,故可用作SPE填料或分子印跡薄膜來分離富集復雜基質中的痕量分析物,已被廣泛應用于獸藥殘留的分析[37]。Zhou等[38]以螺旋霉素為模板,制備了對阿奇霉素具有高選擇性的分子印跡聚合物整體式微柱,建立了基于分子印跡聯用液相色譜-串聯質譜檢測豬肉中阿奇霉素殘留量的簡便、高靈敏度的方法。該法克服了復雜基質和雜質的干擾,比傳統固相萃取法具有更好的凈化效果。
3.1.5加壓溶劑萃取法加壓溶劑萃取通過升溫和加壓提高物質的溶解度和溶質擴散效率,從而提高萃取效率。該方法萃取時間短、萃取溶劑用量少、萃取率高、自動化程度高,可應用于食品中大環內酯類藥物殘留的測定。如Juan 等[39]分析了肉類和牛奶中5種大環內酯類藥物殘留,使用不同量的海砂和EDTA研究了提取方法以及固相分散萃取法,并對溶劑、提取時間、溫度和壓力條件進行了優化。將該方法應用于篩查瓦倫西亞社區市場的牛奶和肉類樣品中的大環內酯和林可酰胺殘留,回收率均在70%以上,4個陽性樣本殘留量均高于歐盟立法規定的最大殘留量,為食品安全風險事件提供了可靠的技術保障。
3.1.6在線固相萃取法與傳統固相萃取技術相比,在線固相萃取具有操作簡便、穩定性好、檢出限低、靈敏度高等優勢。張曉光等[40]采用在線固相萃取凈化/液相色譜-串聯質譜方法檢測豬肉中的10種大環內酯類藥物殘留,樣品以乙腈提取,經在線固相萃取柱凈化,甲醇洗脫后,轉移至色譜柱中進行分離,用四極桿質譜檢測。通過對固相萃取參數、液相色譜參數、質譜參數進行優化,有效降低了基質效應;利用在線固相萃取裝置,克服了傳統固相萃取穩定性差、操作繁瑣、成本高、資源消耗大等缺點。該方法實用性強,后期可對檢測品種進行擴充。
3.2 檢測方法
目前有關大環內酯類藥物殘留的檢測技術主要為高效液相色譜法和液相色譜-質譜聯用法(LC-MS),表2列出了部分大環內酯類藥物殘留的分析條件。

表2 肉類食品中大環內酯類藥物的分析條件Table 2 Analysis conditions of macrolide in meat food
(續表2)
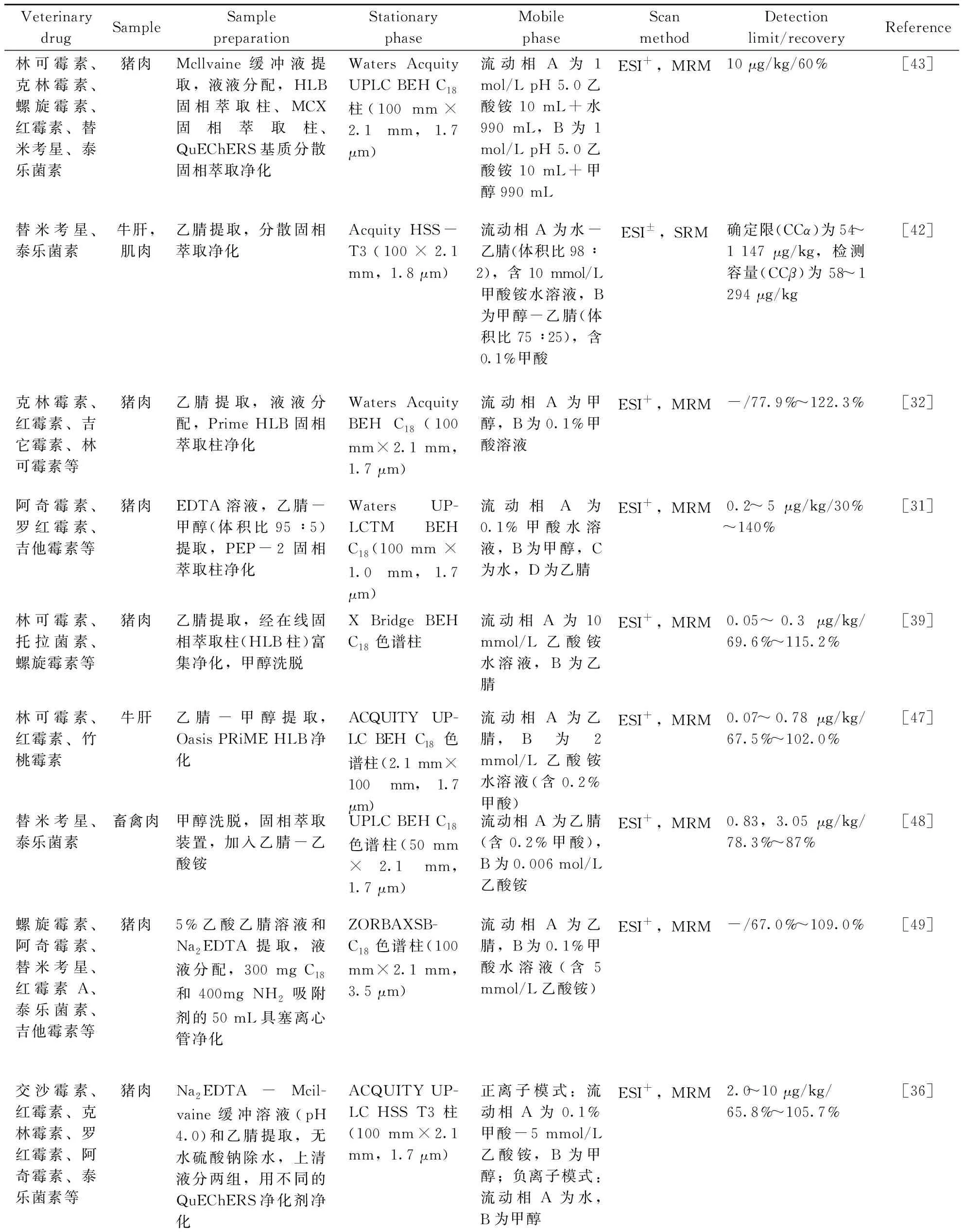
Veterinary drugSampleSample preparationStationary phaseMobile phaseScan methodDetection limit/recoveryReference林可霉素、克林霉素、螺旋霉素、紅霉素、替米考星、泰樂菌素豬肉Mcllvaine緩沖液提取,液液分配,HLB固相萃取柱、MCX固相萃取柱、QuEChERS基質分散固相萃取凈化Waters Acquity UPLC BEH C18柱(100 mm×2.1 mm,1.7 μm)流動相A為1 mol/L pH 5.0乙酸銨10 mL+水990 mL,B為1 mol/L pH 5.0乙酸銨10 mL+甲醇990 mLESI+,MRM10 μg/kg/60%[43]替米考星、泰樂菌素牛肝,肌肉乙腈提取,分散固相萃取凈化Acquity HSS-T3(100×2.1 mm,1.8 μm)流動相A為水-乙腈(體積比98∶2),含10 mmol/L甲酸銨水溶液,B為甲醇-乙腈(體積比75∶25),含0.1%甲酸ESI±,SRM確定限(CCα)為54~1 147 μg/kg,檢測容量(CCβ)為58~1 294 μg/kg[42]克林霉素、紅霉素、吉它霉素、林可霉素等豬肉乙腈提取,液液分配,Prime HLB固相萃取柱凈化Waters Acquity BEH C18(100 mm×2.1 mm,1.7 μm)流動相A為甲醇,B為0.1%甲酸溶液ESI+,MRM-/77.9%~122.3%[32]阿奇霉素、羅紅霉素、吉他霉素等豬肉EDTA 溶液,乙腈-甲醇(體積比95∶5) 提取,PEP-2 固相萃取柱凈化Waters UP-LCTM BEH C18(100 mm × 1.0 mm,1.7 μm)流動相A為0.1%甲酸水溶液,B為甲醇,C為水,D為乙腈ESI+,MRM0.2~5 μg/kg/30%~140%[31]林可霉素、托拉菌素、螺旋霉素等豬肉乙腈提取,經在線固相萃取柱(HLB柱)富集凈化,甲醇洗脫X Bridge BEH C18色譜柱流動相A為10 mmol/L乙酸銨水溶液,B為乙腈ESI+,MRM0.05~0.3 μg/kg/69.6%~115.2%[39]林可霉素、紅霉素、竹桃霉素牛肝乙腈-甲醇提取,Oasis PRiME HLB凈化ACQUITY UP-LC BEH C18色譜柱(2.1 mm×100 mm,1.7 μm)流動相A為乙腈,B為2 mmol/L乙酸銨水溶液(含0.2%甲酸)ESI+,MRM0.07~0.78 μg/kg/67.5%~102.0%[47]替米考星、泰樂菌素畜禽肉甲醇洗脫,固相萃取裝置,加入乙腈-乙酸銨UPLC BEH C18色譜柱(50 mm ×2.1 mm,1.7 μm)流動相A為乙腈(含0.2%甲酸),B為0.006 mol/L乙酸銨ESI+,MRM0.83,3.05 μg/kg/78.3%~87%[48]螺旋霉素、阿奇霉素、替米考星、紅霉素A、泰樂菌素、吉他霉素等豬肉5%乙酸乙腈溶液和Na2EDTA提取,液液分配,300 mg C18和400mg NH2吸附劑的50 mL具塞離心管凈化ZORBAXSB-C18色譜柱(100 mm×2.1 mm,3.5 μm)流動相A為乙腈,B為0.1%甲酸水溶液(含5 mmol/L乙酸銨)ESI+,MRM-/67.0%~109.0%[49]交沙霉素、紅霉素、克林霉素、羅紅霉素、阿奇霉素、泰樂菌素等豬肉Na2EDTA-Mcil-vaine緩沖溶液(pH 4.0)和乙腈提取,無水硫酸鈉除水,上清液分兩組,用不同的QuEChERS凈化劑凈化ACQUITY UP-LC HSS T3柱(100 mm×2.1 mm,1.7 μm)正離子模式:流動相A為0.1%甲酸-5 mmol/L乙酸銨,B為甲醇;負離子模式:流動相A為水,B為甲醇ESI+,MRM2.0~10 μg/kg/65.8%~105.7%[36]

紅霉素、交沙霉素、螺旋霉素、泰樂菌素、竹桃霉素動物組織樣品乙腈提取,液液分配,正己烷脫脂,異丙醇凈化Luna C18 色譜柱(150 mm ×2.0 mm,3 μm)流動相A為0.1%甲酸的乙腈-甲醇(1∶1),B為50 mmol/L乙酸銨水溶液ESI+,MRM0.05~5.3 μg/kg/70.3%~102%[26]
(續表2)

Veterinary drugSampleSample preparationStationary phaseMobile phaseScan methodDetection limit/recoveryReference螺旋霉素、替米考星、阿奇霉素、泰樂菌素、林可霉素、克林霉素動物肌肉0.1%甲酸的乙腈提取,高性能分散儀萃取,離心,在聚丙烯管中低溫純化Agela Durashell RP(100 mm×2.1 mm,5 μm)保護柱填充C18(Security Guard Phe-nomenex,4.0 mm×3.0 mm,5 μm)流動相A為0.1%甲酸水溶液和5 mmol/L甲酸銨,B為含0.1%甲酸和5 mmol/L甲酸銨的甲醇ESI+,SRMCCβ為1.0~50.0 μg/kg,CCα為120.3 μg/L[45]林可霉素、克林霉素、螺旋霉素、替米考星、泰樂菌素等家禽肌肉正己烷脫脂,使用PSA和NH2的管中的分散固相萃取(D-SPE)除雜Acquity HSS-T3色譜柱(100 mm×2.1 mm,1.8 μm)流動相A為0.1%(體積分數)甲酸的甲醇,B為0.1%甲酸ESI+,MRM-/60%~139%[33]替米考星、螺旋霉素、紅霉素A、阿奇霉素等動物肌肉乙酸乙酯提取,液液分配,1 mL 5%氨水的甲醇溶液洗脫,MISPE純化Zorbax SB-Aq C18柱(150 mm×2.1 mm,3.5 μm)流動相A為乙腈,B為0.1%甲酸水ESI+,MRM0.1~0.4 μg/g/60.7%~100.3%[44]多拉菌素、伊維菌素牛肉乙腈提取,C18固相萃取柱凈化C18 柱(250 mm×4.6 mm,5 μm)流動相A為乙腈,B為水熒光檢測器1.5 μg/kg/70.0%~120.0%[41]替米考星、螺旋霉素、紅霉素等牛肉、豬肉、雞肉乙腈、甲酸銨提取,分散固相萃取凈化YMC-Triart C18柱(3.0 mm×100 mm,3 μm)流動相A為水(含0.1%甲酸),B為乙腈(含0.1%甲酸)ESI-,MRMCCβ為17~358 μg/kg/60%~217%[50]
-not reported
3.2.1液相色譜法高效液相色譜適用于極性大、沸點高的化合物分離分析。該法采用一定的提取手段將被分析對象溶于可作為流動相的溶劑中,直接進行檢測分析,具有柱效高、選擇性好、價格低、分析速度快、應用范圍廣等優勢,被廣泛應用于食品中獸藥殘留的檢測。根據食品基質的不同選擇不同的檢測器,常用的檢測器有紫外檢測器(UVD)、熒光檢測器(FLD)、電化學檢測器(ECD)、二極管陣列檢測器(DAD)等。由于MALs難以氣化,并且多具有強的紫外線吸收,所以高效液相色譜法是MALs殘留的常用測定方法。王梅等[41]采用高效液相色譜法/熒光檢測器測定牛肉組織中多拉菌素、伊維菌素殘留量,兩種待測物在3.0~200.0 μg/L 范圍內呈良好的線性關系,樣品加標回收率為70.0%~120.0%,方法檢出限為1.5 μg/kg。該方法有效簡化了現行農業部781號公告標準的繁瑣處理步驟,縮短了衍生時間,有效保護了色譜柱,且操作簡單,重復性好,但只能用于已知目標化合物的分析,無法進行未知化合物的篩查。
3.2.2液相色譜-質譜聯用法液相色譜-質譜聯用法(LC-MS)在獸藥分析領域的應用日益廣泛,因能夠以較高強度提供更窄的峰值,從而可顯著提高方法的靈敏度和選擇性,并可加快分析速度,具有較寬的線性范圍,適合于確證分析,故被用于大環內酯類藥物的殘留分析[42]。LC-MS除可分析強極性、難揮發、熱不穩定的化合物之外,還具有分析范圍廣、分離能力強、定性分析結果可靠、檢出限低、分析時間快、自動化程度高等優點。目前各種食品基質中大環內酯類藥物的液質聯用檢測方法均有報道。
高分辨飛行時間質譜(Time of flight mass spectrometry,TOF MS)近年來在食品安全分析領域的應用也越來越廣泛,與低分辨的串聯四極桿質譜相比,TOF MS具有定性與定量結果準確、靈敏度高、質量精度高、具有正負切換掃描功能等優勢。郝杰等[43]使用高分辨飛行時間質譜對豬肉中包括大環內酯類在內的29種抗菌藥物進行同時檢測,并比較了不同固相萃取柱的凈化效果,選用HLB固相萃取柱,建立了基于精確質量數的篩查數據庫和優化的定量方法,29種抗菌藥物的定量下限為0.5~10 μg/kg,平均回收率為50.10%~118.33%。該方法滿足食品安全風險突發事件快速應急處理工作中高通量、高可靠性確證和定量分析的要求,為相關工作提供了可靠的技術保障。
Song等[44]以泰樂菌素作為模板,甲基丙烯酸作為功能單體合成分子印跡聚合物(MIPs),采用分子印跡固相萃取與液相色譜-串聯質譜聯用的方法測定動物肌肉樣品中10種大環內酯類藥物殘留。結果表明,MIP對大環內酯類藥物具有良好的識別性能,比傳統固相萃取具有更好的凈化效果。藥物的回收率為60.7%~100.3%,檢出限為0.1~0.4 μg/kg,該方法可用于動物肌肉中大環內酯類藥物殘留的常規監測。Jank等[45]建立了肉類食品中大環內酯類藥物殘留的篩查方法,該方法采用2002/657/EC標準進行了驗證,確定了特異性驗證參數,檢測容量(CCβ)為1.0~50.0 μg/L,確定限(CCα)為120.3 μg/L。該方法快速、簡便,適用于常規實驗室的高通量分析,已應用于巴西國家殘留控制計劃,用于抗生素殘留確定。
4 結 論
近年來我國在獸藥殘留檢測領域發展迅速,對藥物殘留的檢測能力也大大提升。但隨著藥物結構的逐漸復雜化、低劑量化和新型藥物研發數量的日益增多,對檢測技術的要求也越來越高。然而國內關于快速檢測大環內酯類藥物及其代謝產物的相關研究尚不完善,有待于進一步開發探索,以實現快速準確地從復雜基質中分離大環內酯類藥物及其代謝產物,并獲得較高的回收率和較好的測定結果。如何高效地將大環內酯類藥物及代謝產物從復雜基質中分離,并建立動物源性食品中多類獸藥殘留及其代謝產物的非定向篩查方法,將成為新的研究趨勢。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
