硫熏及不同硫熏程度對白術化學成分的影響△
2018-11-06 05:58:22徐春良付輝政趙雯戴冕任琦鐘瑞建
中國現代中藥 2018年10期
徐春良,付輝政,趙雯,戴冕,任琦*,鐘瑞建*
(1.江西中醫(yī)藥大學附屬醫(yī)院,江西 南昌 330006;2.江西江中中藥飲片有限公司,江西 九江 332300;3.江西省藥品檢驗檢測研究院/江西省藥品與醫(yī)療器械質量工程技術研究中心,江西 南昌 330029)
白術為菊科植物白術Atractylodes macrocephala Koidz.的干燥根莖,具有健脾益氣,燥濕利水,止汗,安胎之功效。用于脾虛食少,腹脹泄瀉,痰飲眩悸,水腫,自汗,胎動不安[1-4]。白術始載于 《神農本草經》,是常用大宗藥材,俗有 “南術北參、十方九術”之說,為健脾補氣第一要藥,廣泛應用于臨床[5]。白術含有較多揮發(fā)性成分、內酯類成分、苷類及多糖類成分[6-7],儲存不當容易發(fā)生 “走油變色”,且影響其外觀和質量。白術在產地加工中,常采用硫磺熏蒸的方法進行干燥,經硫磺熏蒸后白術外觀性狀更加美觀不易變色且更易保存[8]。《中華人民共和國藥典》2015年版一部規(guī)定白術藥材及飲片中二氧化硫殘留量不得大于 400 mg·kg-1[1],筆者在2017年白術國家評價性抽驗發(fā)現,部分白術飲片二氧化硫殘留量超過規(guī)定限度1倍以上。硫熏不僅影響中藥藥效及安全性,還可能會破壞藥材化學成分[9-11]。目前未見不同硫磺熏蒸程度對白術化學成分的影響研究。本實驗首先采用薄層色譜法篩查硫熏對白術化學成分的變化,通過硅膠柱色譜分離制備和波譜技術分析確證發(fā)生變化的化學成分結構,其次采用離子色譜法和高效液相色譜法考察不同硫磺熏蒸程度白術與發(fā)生變化化學成分的相關性,為白術飲片的科學炮制、合理應用及質量評價提供參考。
1 儀器與試藥
1.1 儀器
Thermo Fisher ScientificICS-1600離子色譜儀、淋洗液自動發(fā)生器、陰離子交換柱;CAMAG TLC Visualizer薄層色譜數碼成像系統;Waters Xevo G2QTOF飛行時間質譜儀;SHIMADAU LC-20AD高效液相色譜儀,四元泵、自動進樣器、柱溫箱、PAD檢測器、LC solution工作站;Sartorius BT 25 S十萬分之一電子天平。
1.2 試藥
東莨菪內酯對照品(中國食品藥品檢定研究院,批號:110768-200504,供含量測定用);硅膠G板(青島海洋化工廠分廠);乙腈、甲醇為色譜純,水為娃哈哈飲用純凈水,其他試劑均為分析純。
白術(批號:20160901、170110、170209)購于浙江磐安藥材市場,經江西省藥品檢驗檢測研究院付輝政副研究員鑒定為菊科植物白術 Atractylodes macrocephala Koidz.的干燥根莖。
2 方法與結果
2.1 硫磺熏蒸白術的制備
取白術飲片,采用有機玻璃材料自制熏箱進行硫磺熏蒸。熏箱內設一層網狀隔板,將200 g白術飲片均勻擺放在隔板上,將定量硫磺(0.5、1、2、3 g)置于坩堝內并點燃形成藍色火焰時放置于熏箱底部,熏至24 h后將白術飲片放置于恒溫干燥箱80℃干燥。
2.2 硫磺熏蒸對白術化學成分的影響
2.2.1 薄層色譜定性篩選 取同一批硫熏與未硫熏白術樣品粉末各4 g,加甲醇20 mL,超聲處理45 min,濾過,濾液作為供試品溶液。吸取上述供試品溶液20μL,分別點于同一硅膠G薄層板上,以三氯甲烷-丙酮(19∶1)為展開劑,展開,取出,晾干,在紫外光燈(365 nm)下檢視。結果見圖1。

圖1 白術樣品中香豆素類成分薄層色譜圖
由圖1可知,白術經硫熏后,斑點1和2含量明顯下降,說明硫熏會對白術化學成分產生影響。取不同批次的樣品3批,同法實驗,結果一致。為明確斑點1和2的結構,進一步對斑點1和2進行了制備及結構確證。
2.2.2 目標產物的制備 取白術(2.5 kg),粉碎,用10倍量80%乙醇回流提取3次,每次2 h。提取液濃縮至適量,用乙酸乙酯萃取4次,萃取液減壓濃縮至干,得乙酸乙酯部分(105 g)。將乙酸乙酯部分與硅膠拌樣,經正相硅膠柱色譜分離,以三氯甲烷-丙酮(50∶1~10∶1)梯度洗脫,經薄層色譜檢視,合并相近的組分,得 10個組分(Fr.1~Fr.10)。Fr.6(8.6 g)以三氯甲烷-丙酮(15∶1)等度洗脫,得到目標產物1(斑點1)和2(斑點2)。
2.2.3 目標產物結構鑒定 目標產物1:淡黃色針狀結晶(甲醇)。ESI-MS m/z:193[M+H]+,191[M-H]-。1H-NMR(600 MHz,CD3OD)δ:7.83(1H,d,J=9.4 Hz,H-4),7.08(1H,s,H-8),6.71(1H,s,H-5),6.18(1H,d,J=9.4 Hz,H-3),4.88(1H,s,-OH),3.88(3H,s,-OCH3);13C-NMR(150 MHz,CD3OD)δ:164.1(C-2),112.5(C-3),146.2(C-4),109.3(C-5),146.8(C-6),153.3(C-7),103.6(C-8),151.6(C-9),112.3(C-10),56.8(OCH3)。綜合上述信息,將以上波譜數據與文獻數據對照基本一致[12],鑒定為東莨菪內酯。
目標產物2:白色針狀結晶(甲醇)。ESI-MS m/z:163[M+H]+,161[M-H]-。1H-NMR(600 MHz,CD3OD)δ:7.81(1H,d,J=9.4 Hz,H-4),7.41(1H,d,J=8.5 Hz,H-5),6.16(1H,d,J=9.4 Hz,H-3),6.75(1H,dd,J=8.5,2.2 Hz,H-6),6.70(1H,d,J=2.2 Hz,H-8);13C-NMR(150 MHz,CD3OD)δ:163.5(C-2),113.8(C-3),146.1(C-4),130.4(C-5),113.2(C-6),161.9(C-7),103.3(C-8),157.8(C-9),112.1(C-10)。綜合上述信息,將以上波譜數據與文獻數據對照基本一致[13],鑒定為傘形花內酯。
3 不同劑量硫磺熏蒸對白術中東莨菪內酯的影響
3.1 東莨菪內酯的HPLC含量測定
3.1.1 色譜條件 色譜柱為Inertsil ODS-SPC18(250 mm×4.6 mm,5μm),流動相為乙腈-水(8∶92),檢測波長為344 nm,柱溫為30℃,進樣量為20μL。在上述色譜條件下,東莨菪內酯分離情況及峰形良好。色譜圖見圖2。
3.1.2 對照品溶液的制備 取東莨菪內酯對照品適量,精密稱定,加甲醇制成每l mL含4μg的溶液,即得。
3.1.3 供試品溶液的制備 取本品粉末(過四號篩)約4 g,精密稱定,置具塞錐形瓶中,精密加入甲醇20 mL,密塞,稱定質量,超聲處理(功率500 W,頻率40 kHz)45 min,放冷,再稱定質量,用甲醇補足減失的質量,搖勻,濾過,取續(xù)濾液,即得。
3.1.4 線性關系考察 精密稱取東莨菪內酯對照品10.26 mg,置50 mL量瓶中,加甲醇溶解并稀釋至刻度,搖勻,得東莨菪內酯對照品溶液。吸取東莨菪內酯對照品溶液,加甲醇分別稀釋至質量濃度為1.026、2.052、4.104、6.146、8.208μg·mL-1,分別吸取20μL按上述色譜條件測定峰面積。以進樣量(X)為橫坐標,峰面積(Y)為縱坐標,繪制標準曲線,得回歸方程為Y=3.693 1×106X-3.074 0×103(r=0.999 9),東莨菪內酯進樣量在0.020 5~0.164 2μg與峰面積呈良好線性關系。
3.1.5 精密度試驗 精密吸取按3.1.2制備的對照品溶液20μL,按3.1.1色譜條件連續(xù)進樣6次,測定峰面積。結果東莨菪內酯峰面積的RSD=0.5%(n=6),表明儀器精密度良好。
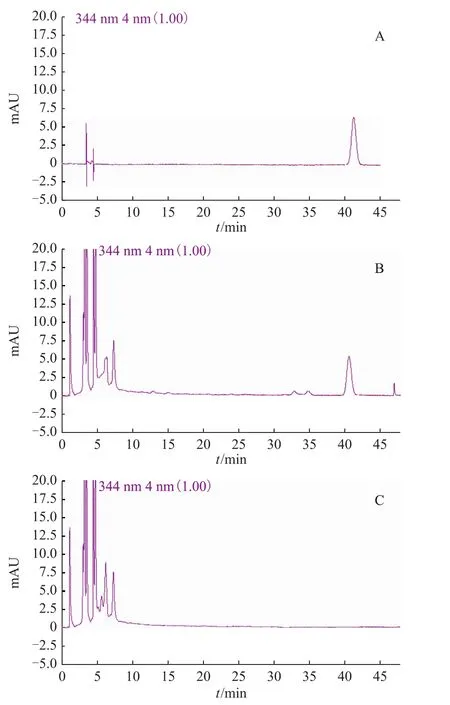
圖2 白術及對照品HPLC圖
3.1.6 重現性試驗 取樣品(批號:170110)6份,每份約4 g,精密稱定,按3.1.3制備供試品溶液,按3.1.1色譜條件進行分析測定,東莨菪內酯含量的平均值為13.601 2 mg·kg-1,RSD=1.7%,表明該方法重現性良好。
3.1.7 穩(wěn)定性試驗 精密吸取供試品溶液(批號:170110)20μL,分別于 0、5、10、15、20、24 h進樣測定,測得東莨菪內酯峰面積的RSD=1.6%(n=6),說明供試品溶液在24 h內穩(wěn)定。
3.1.8 回收率試驗 采用加樣回收率法,精密稱取已知含量(批號:170110)的樣品 6份,每份約2.0 g,精密稱定,置具塞錐形瓶中,每份精密加入含東莨菪內酯1.351 0μg·mL-1對照品溶液20 mL,按3.1.3制備供試品溶液,按3.1.1色譜條件進行分析測定,計算回收率。結果東莨菪內酯的平均回收率為100.6%(n=6),RSD=0.7%。見表1。
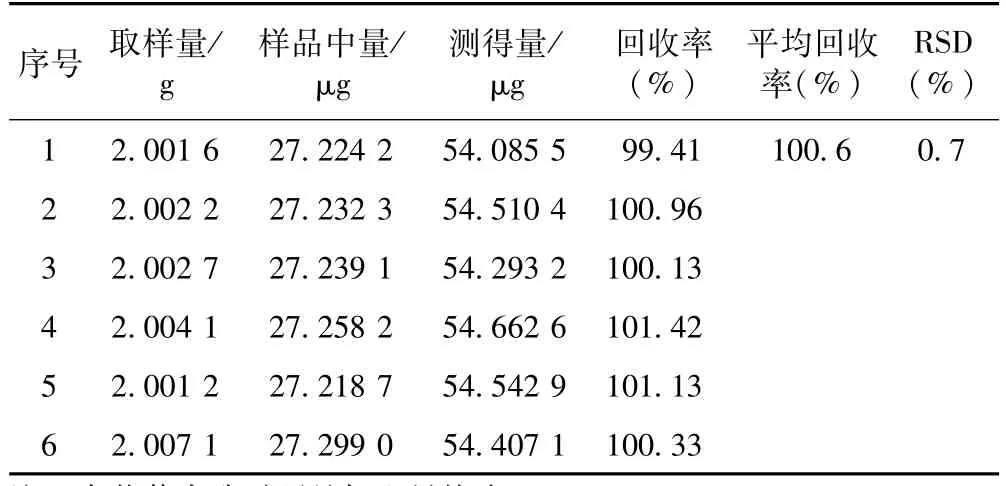
表1 白術中東莨菪內酯回收率試驗結果(n=6)
3.1.9 耐用性試驗 取樣品按3.1.3制備供試品溶液,分別用 Inertsil ODS-SP C18(250 mm×4.6 mm,5μm)、Agilent XDB-C18(250 mm×4.6 mm,5μm)和Waters Sunfire C18(250 mm×4.6 mm,5μm)3種品牌的色譜柱按3.1.1色譜條件進行測定,含量測定結果無明顯差別。
3.1.10 樣品測定 取白術樣品15批,每個樣品取3份,按3.1.3制備供試品溶液。精密吸取供試品溶液20μL,按3.1.1色譜條件進樣分析,測定峰面積,用外標法計算出東莨菪內酯的含量,測定結果見表2。
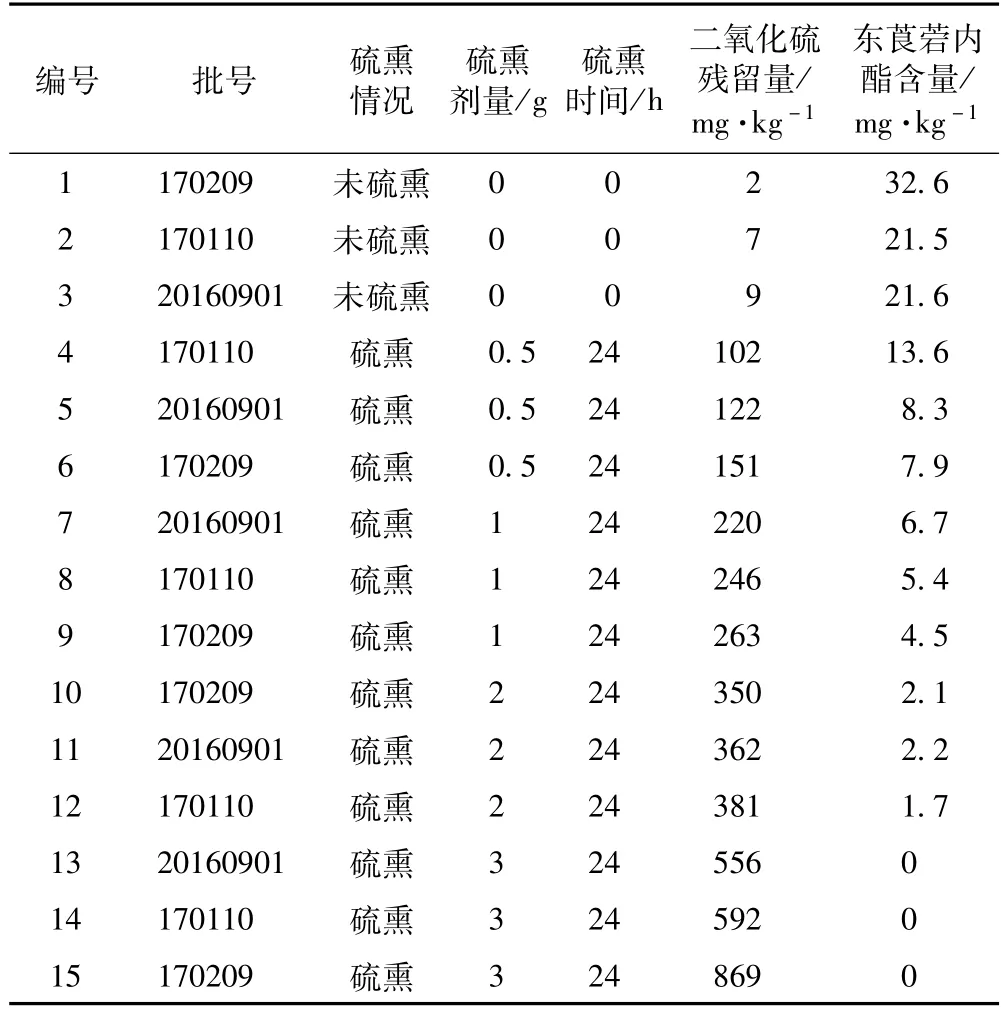
表2 白術中二氧化硫殘留量及東莨菪內酯含量測定結果(n=3)
3.2 二氧化硫殘留量測定
3.2.1 色譜條件 陰離子交換柱(AS11-HC,250 mm×4 mm),淋洗液為20 mmol·L-1氫氧化鉀溶液,抑制電流為 50 mA,流速為 1.0 mL·min-1,柱溫為30℃,進樣量為25μL[14-15]。二氧化硫殘留量測定離子色譜圖見圖3。
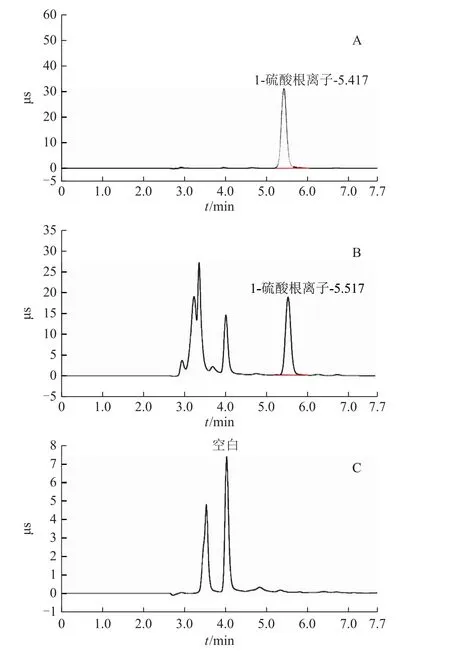
圖3 白術中二氧化硫殘留量測定離子色譜圖
3.2.2 對照品溶液的制備 精密吸取硫酸根標準溶液(1000μg·mL-1),加水制成1、5、20、50、100、200μg·mL-1的溶液,繪制標準曲線。以進樣量(X)為橫坐標,峰面積積分值(Y)為縱坐標,繪制標準曲線,得回歸方程為Y=0.237 8 X-0.036 1(r=0.999 9),硫酸根在 0.025~5.000μg與峰面積呈良好線性關系。
3.2.3 供試品溶液的制備 取白術粉末(過四號篩)約5 g,精密稱定,置瓶 A(兩頸燒瓶)中,加水50 mL,振搖,使分散均勻,接通水蒸氣蒸餾瓶。吸收瓶B(100 mL量瓶)中加入3%過氧化氫溶液20 mL作為吸收液,吸收管下端插入吸收液液面以下。A瓶中沿瓶壁加入5 mL鹽酸,迅速密塞,開始蒸餾,保持C瓶沸騰并調整蒸餾火力,使吸收管端的餾出液的流出速率約為2 mL·min-1。蒸餾至瓶B中溶液總體積約為95 mL,用水洗滌尾接管并將其轉移至吸收瓶中,并稀釋至刻度,搖勻,放置1 h后,以0.45μm微孔濾膜濾過,即得。
3.2.4 方法學考察 取對照品溶液測定6次,進行精密度考察;取供試品溶液,分別于0、5、10、15、20、24 h測定,進行穩(wěn)定性考察;另取白術粉末(過四號篩)約5 g,共6份,按3.2.3制備并測定,進行重復性考察;再取白術粉末(過四號篩)約2.5 g,共6份,分別精密加入一定量的亞硫酸鈉對照品,按3.2.3測定,進行加樣回收率考察。結果表明,精密度、穩(wěn)定性、重復性、加樣回收率測定結果的RSD值均在2.0%以內;樣品溶液二氧化硫殘留量平均質量濃度為 220 mg·mL-1,24 h內測定穩(wěn)定,平均加樣回收率為97.52%。
3.2.5 樣品測定 分別取白術粉末(過四號篩)15批,按3.2.3制備供試品溶液。精密吸取供試品溶液25μL,按3.2.1色譜條件進樣,測定,計算樣品中的硫酸根含量,按照(SO2/SO42-=0.666 9)計算樣品中二氧化硫的含量。測定結果見表2。
4 討論
本實驗采用薄層色譜法對硫熏前后白術中化學成分進行定性篩選,并通過硅膠柱色譜分離制備和波譜數據分析確證硫熏對白術中產生影響的化學成分的結構。為了進一步考察硫黃熏蒸程度對白術中東莨菪內酯和傘形花內酯的相關性。由于白術中傘形花內酯含量非常低且液相色譜分離效果不佳,本次實驗僅以東莨菪內酯為指標,采用離子色譜法和高效液相色譜法分別對白術中二氧化硫殘留量和東莨菪內酯的含量進行測定,其結果重復性良好,方法可行。
實驗結果表明,硫熏對白術中香豆素類成分東莨菪內酯和傘形花內酯有顯著的影響,其中東莨菪內酯含量隨著二氧化硫殘留量增加,整體呈下降趨勢直至為零。為了避免活性成分的下降,保證藥材及飲片的質量,進而確保臨床療效,建議研究白術合理適量硫磺熏蒸的加工方法,并制定合理的二氧化硫殘留量限度。
