硝酸異丙酯水解反應的量子化學計算
2018-11-12 03:37:54陳永康安振濤陳明華
火炸藥學報 2018年5期
陳永康,安振濤,陳明華,張 力
(1.陸軍工程大學石家莊校區,河北 石家莊 050003;2. 特種勤務研究所,河北 石家莊 050003)
引 言
硝酸異丙酯是一種硝酸酯類的含能材料,不僅可以作為戰斗部裝藥,也可用于火箭推進劑。除燃爆特性以外,它還具有強氧化性、強揮發性和毒性,廢棄的硝酸異丙酯需要及時進行處理,否則將造成嚴重的安全隱患。而當前還沒有針對硝酸異丙酯的安全快速去能處理方法,通常是采用揮發、燒毀、炸毀等方式處理,存在環境污染嚴重、危險性大、資源浪費等問題,不符合當前“綠色環保、安全高效”的要求,因此尋找安全綠色的硝酸異丙酯處理方法是亟待解決的問題。
根據硝酸異丙酯屬于酯類的特點,對其進行水解是一種相對有效的處理方法。目前,關于硝酸異丙酯性質和水解反應機理已有一定的理論研究[1-6]。曾秀琳等[7]采用密度泛函方法計算了其前線軌道能并分析了其對熱穩定性的影響;True等[8]通過實驗探索了硝酸異丙酯的構型;Thornton[9]根據經典力學對其水解機理做出了理論推斷;貢雪東等[10]運用SCF-MOAM1方法確定了硝酸異丙酯的幾何構型,并通過AM1方法得到了氣相狀態下硝酸異丙酯的SN2堿性水解位能曲線;劉志勇等[11]分別用AM1和B3LYP/6-31+G(d)計算了氣相狀態下硝酸異丙酯的熱分解初始反應機理和水解反應機理。
本研究在此基礎上,采用更高精度的計算方法,考慮其溶劑化效應及pH值的影響,計算了反應動力學,對硝酸異丙酯的水解機理進行研究。
1 計算方法
所有的電子結構和能量計算均由Guassain 09程序包完成。采用從頭算二階多體微擾理論(second order Mφller-Pleset perturbation theory, MP2) 6-311+g(d,p)基組優化各反應中反應物、產物和過渡態的幾何結構并計算振動頻率,MP2方法要求在Hartree-Fock計算之后接著進行Mφller-Pleset關聯能校正計算,對MP2截斷到二階。對反應過渡態進行內稟反應坐標(IRC)計算以對過渡態加以確認。反應體系為帶電體系,對體系進行閉殼層計算。采用極化連續介質模型(Polarizable Continuum Model, PCM)研究了反應的溶劑化效應。在MP2/6-311+g(d,p)水平下計算得到標準態和溶劑化條件下的反應能級圖。為確定pH值對水解反應的影響,在MP2/6-311+g(d,p)水平計算得到不同pH值時反應物、產物、過渡態及絡合物的能量。
反應的動力學計算由Polyrate 2015程序完成。在CCSD(T)/6-311++g(3df,2p)水平下,通過經典過渡態理論 (TST),正則變分過渡態理論(CVT),零曲率隧道效應校正的正則變分過渡態理論 (CVT/ZCT)和小曲率隧道效應校正的正則變分過渡態理論 (CVT/SCT) 對反應速率常數進行計算。
2 結果與討論
2.1 反應路徑
對親核取代SN2反應和β-H消除Eβ反應這兩種反應路徑進行了計算比較,兩種反應路徑如圖1所示。親核取代反應(以下稱為R1)的產物為異丙醇和硝酸根離子,β-H消除反應(以下稱為R2)的產物為丙烯,硝酸根離子以及水。
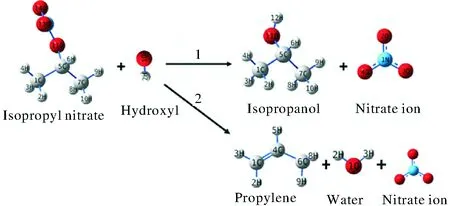
圖1 硝酸異丙酯堿性水解反應Fig.1 The alkaline hydrolysis reaction of isopropyl nitrate
結合圖1,根據硝酸異丙酯的幾何結構可以發現,氫氧根離子進攻中間碳原子僅有一種路徑,而氫氧根進攻末端氫原子時,由于氫原子不具有對稱性,因此有3種路徑。即本研究中的硝酸異丙酯堿性水解過程共包括4條反應路徑:
Pathway 1:氫氧根離子從硝酸異丙酯背面進攻中間碳原子,發生SN2親核取代反應,反應產物為異丙醇和硝酸根離子。
Pathway 2:氫氧根離子進攻硝酸異丙酯末端碳上的9號氫原子,生成丙烯、水、硝酸根離子。
Pathway 3:氫氧根離子進攻硝酸異丙酯末端碳上的10號氫原子,生成丙烯、水、硝酸根離子。
Pathway 4:氫氧根離子進攻硝酸異丙酯末端碳上的8號氫原子,生成丙烯、水、硝酸根離子。
2.2 幾何構型
根據以上4種反應路徑,確定各路徑的過渡態,然后在MP2/6-311+g(d,p)水平下優化各路徑的反應物、過渡態及產物的幾何結構,如圖2所示。
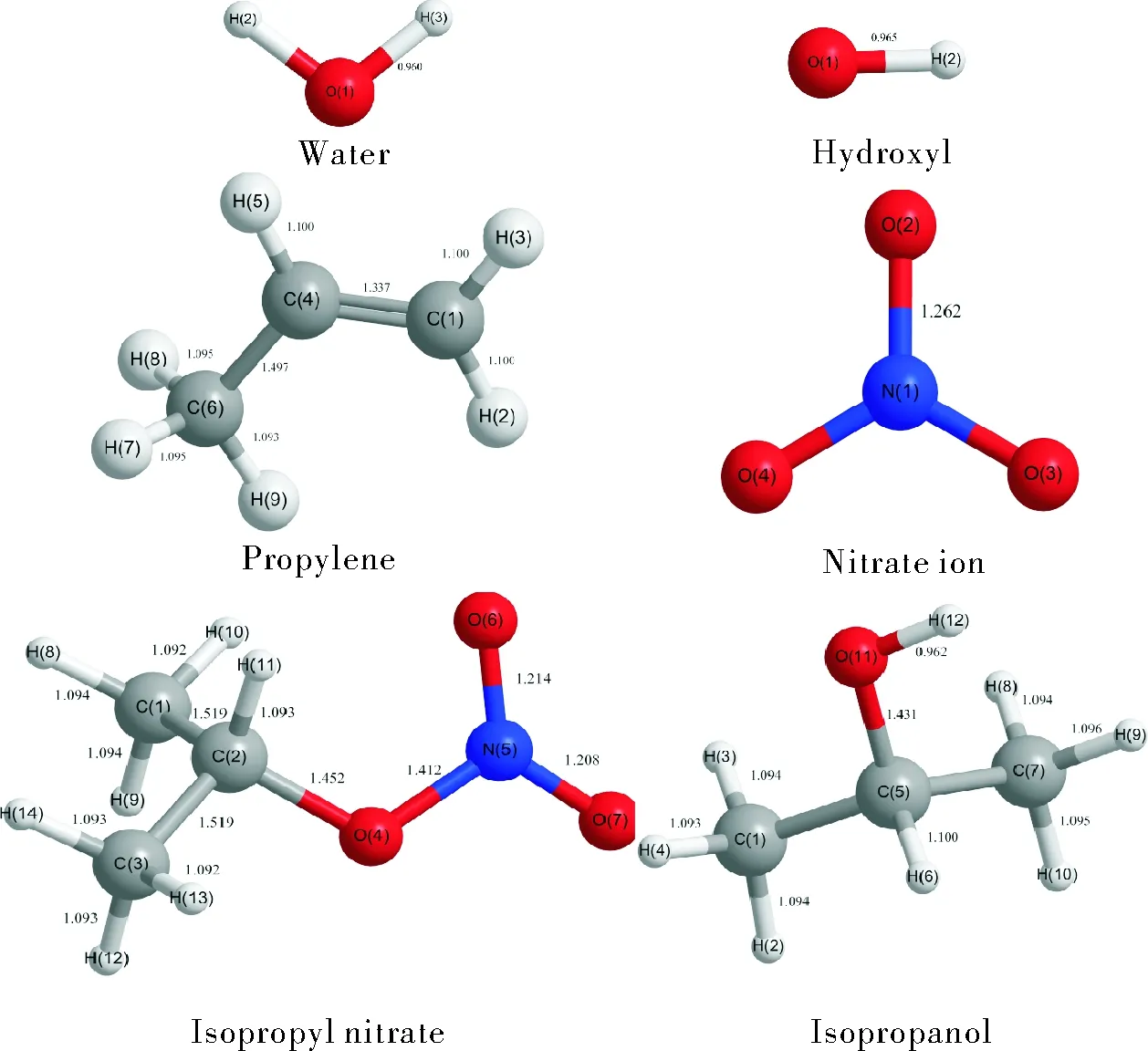
圖2 所有反應物和產物的幾何結構Fig.2 Geometries of all reactants and products
對于硝酸異丙酯的構型,研究表明,O—NO2具有平面結構,C—O—NO2基本共面,硝酸酯基本不可能繞O—NO2旋轉。同時也普遍認為硝酸異丙酯為反式構型,但與硝酸正丙酯不同的是,由于支鏈的影響,硝酸異丙酯中的C—O—NO2并不像硝酸正丙酯一樣完全共面,其C5O11N12O14二面角約為178°。
結合駐點結構,對于兩種反應,在R1中,氫氧根從背面進攻中間的碳原子C5,氫氧根的O逐漸靠近C5,首先形成絡合物1, C5—O11鍵伸長,形成過渡態;然后氫氧根失電子與C5結合,C5—O11鍵斷裂,O11得電子,形成絡合物2;最后,絡合物2分解生成硝酸根離子和異丙醇。在R2中,氫氧根離子進攻硝酸異丙酯末端碳上的氫原子,首先形成絡合物1,氫氧根的O逐漸靠近末端氫原子,C5—O11鍵伸長,形成過渡態;而后末端碳原子的C-H鍵斷裂,失去的氫原子與氫氧根結合,末端碳原子與C5形成碳碳雙鍵,C5—O11鍵斷裂,O11得電子,形成絡合物2;最終絡合物2分解生成丙烯、水和硝酸根離子。
2.2 前線軌道能和頻率計算
在同水平下計算得到3種物質的前線軌道能,最低未占分子軌道能(ELUMO)和最高已占分子軌道能(EHOMO),并計算能級差(ΔE)。其中ΔE=ELUMO-EHOMO,結果見表1。一般認為,能級差越小,電子越容易從最高已占分子軌道向最低未占分子軌道躍遷,分子就越不穩定。
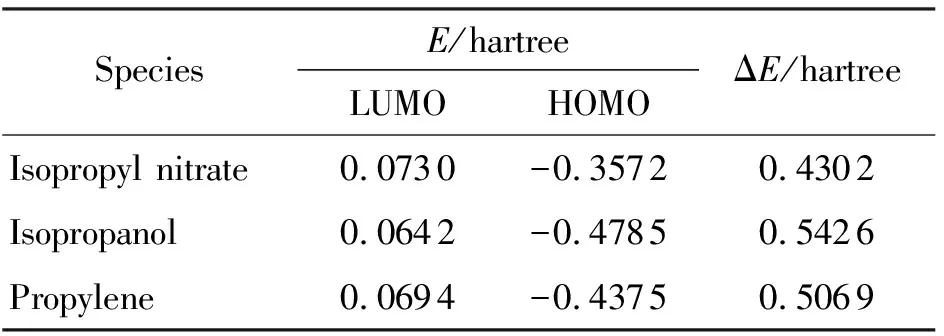
表1 主要反應物和產物的前線軌道能
根據表1中的結果,反應物硝酸異丙酯的能級差最小,兩種產物中異丙醇的能級差高于丙烯,一定程度上說明異丙醇比丙烯更加穩定。曾秀琳等[7]采用密度泛函方法B3P86/6-31G*計算得到硝酸異丙酯的能級差(ΔE)為0.2508hartree,比本研究MP2方法的計算結果小,其原因可能是B3P86方法計算了更多的電子相關、電子排斥效應,因此所得結果更小。
在同水平下計算各反應物、產物及過渡態的振動頻率,反應物和產物的頻率均為正值。異丙醇的O—H伸縮振動頻率的計算值和實驗值分別為3879和3819cm-1;氫氧根振動頻率的計算值和實驗值分別為3269和3309cm-1,計算結果與相關的實驗值較為吻合。過渡態有且僅有一個虛頻。通道1的過渡態TS1的虛頻為600i,通道2~4的過渡態TS2、TS3、TS4的虛頻相互接近,分別為1101i、1154i、1107i。
2.3 溶劑化效應
水解反應實際是在溶液中進行的,分子和過渡態在氣態和在溶液中的性質會存在較大區別,溶劑化效應會對分子能量和反應能壘等造成影響[12-16]。而目前尚未見關于該反應溶劑化效應的研究報道,因此本研究采用極化連續介質模型(PCM)對反應的溶劑化效應進行研究。
在MP2/6-311+g(d,p)水平下對各反應物和產物在標準態和溶劑化條件下的能量同時進行計算,得到所有反應物和產物在標準態和溶劑化條件下的結構能量(E)和零點能校正(ZPE),結果見表2。計算得到兩種反應在標準態和溶劑化條件下的反應焓變(ΔH),結果見表3。
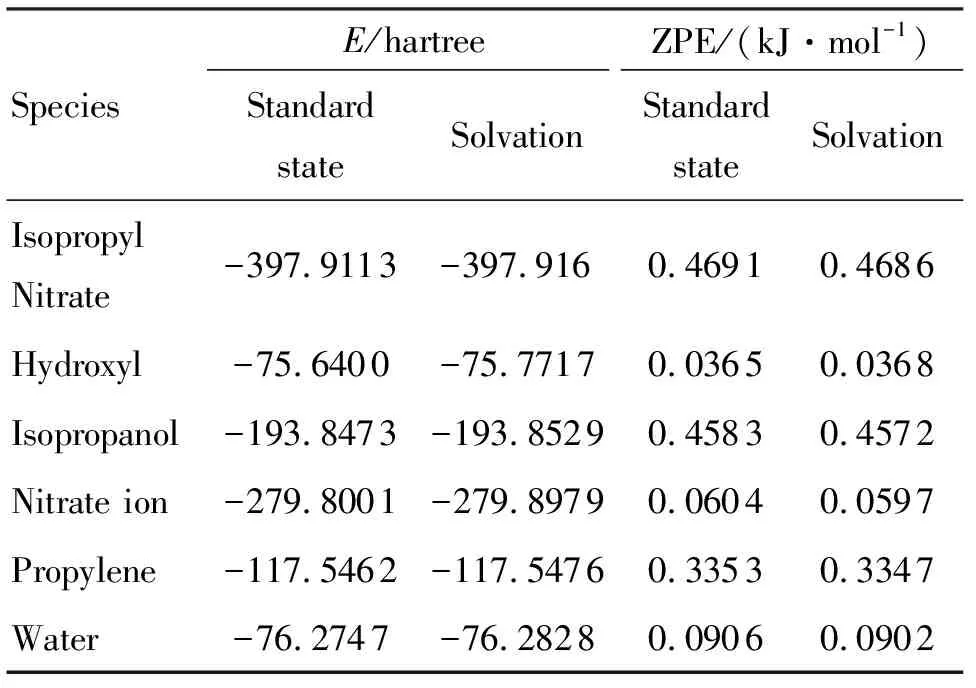
表2 所有反應物和產物的熱力學數據

表3 R1與R2的反應焓變
由表3可知,溶劑化條件下兩種反應的反應焓比標準態高約80kJ/mol,另外,兩種條件下R2的反應焓都要高于R1。文獻[11]計算得到的標準態下R1的理論值與本研究計算結果接近。
圖3為MP2/6-311+g(d,p)水平下計算得到的經零點能校正后標準態和溶劑化條件下的反應能級示意圖,各駐點能量按圖例順序列出。
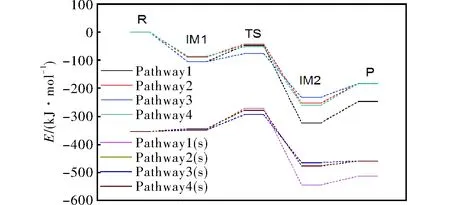
圖3 標準態和溶劑化條件下的反應能級示意圖Fig.3 Schematic diagram of reaction level under standard state and solvation conditions
由圖3可知,在標準態下,反應物首先生成中間體IM1,IM1經由過渡態生成IM2,這一步Pathway1至Pathway4的能量差分別為55.86、45.61、27.91、33.85kJ/mol,最后IM2越過能壘生成產物。溶劑化條件下反應的能壘順序與標準態下基本一致。但不同的是在溶劑化條件下,反應勢能曲線的能量遠低于標準態下反應勢能曲線的能量。
在硝酸異丙酯水解的所有反應路徑當中,Pathway 3(s)的過渡態能壘最低。標準態下,Pathway 2過渡態能壘最高,Pathway 3過渡態能壘最低;溶劑化條件下,Pathway 1(s)過渡態能壘最高,為82.92kJ/mol,Pathway 3(s)過渡態能壘最低,為60.04kJ/mol,這意味著在低溫下,反應易朝著Pathway 3(s)的方向進行。然而對比產物能量可知,Pathway 1(s)的產物能量最低,說明在高溫下,當能量足夠越過反應能壘時,反應傾向于Pathway 1(s)方向,生成穩定的異丙醇和水。
貢雪東等[10]通過半經驗AM1方法求得氣相條件下R1的活化能為75.73kJ/mol;劉志勇等[11]用B3LYP/6-31+G(d)計算了氣相狀態下R1的活化能為52.51kJ/mol。本研究計算得到的R1標準態下的過渡態能壘比文獻值高,意味著水解反應在實際情況下可能不容易進行。
2.4 pH值對水解反應的影響
為確定溶液pH值對水解反應的影響,在MP2/6-311+g(d,p)水平計算得到不同pH值時反應物、產物、過渡態及絡合物的能量,如表4所示。從表4中可以看出,當pH值為8~12時,各駐點能量基本不變,當pH值繼續變大達到13、14時,駐點能量略有升高,總之,pH值的變化對反應物、絡合物1、過渡態、絡合物2、以及產物的能量影響不大,該計算結果表明,pH值不影響反應路徑順序。其原因可能是pH值的變化主要影響水解反應的反應速率,對能量的影響較小。

表4 不同pH值下所有反應物(R)、過渡態(TS)、絡合物(IM1和IM2)和產物(P)的能量
2.5 動力學計算
在CCSD(T)/6-311++g(3df,2p)水平,氣態條件下,通過TST、CVT、CVT/ZCT和CVT/SCT理論方法[17-19]對273~373K溫度范圍內R1的反應速率常數進行計算,得到的反應勢能面如圖4所示,4種方法計算出的反應速率常數隨1000/T的變化曲線見圖5。
由圖4和圖5可知,隨著溫度的升高,反應速率常數逐漸增大。但總體而言,計算得到的該反應的速率常數較小,表明在實際情況下,反應速率較慢。同時,MP2方法計算速率常數值比實際情況偏小,可能會導致計算結果較小。

圖4 R1的反應勢能面Fig.4 Potential energy surface of R1

圖5 R1的反應速率常數隨1000/T的變化曲線Fig.5 Reaction rate constants vs. 1000/T curves of R1
另外,TST和CVT 方法的計算值接近,這意味著整個過程中的變分效應很小,所得結果準確。TST、CVT方法的速率常數計算結果與CVT/ZCT、CVT/SCT方法相比,在低溫處基本一致,在高溫處略有偏差,表明在高溫處的隧道效應對計算結果有一定影響。
在273~373K范圍內,假定活化能不隨溫度發生改變,根據Polyrate的計算數據,對lnk和1/T進行線性擬合,得到反應速率常數k的Arrhenius表達式為:k=1.18×10-23exp(-7357/T),k單位為cm3/(molecule·s)。同時,對原始數據進行非線性擬合,得到三參數的Arrhenius表達式為:k=9.14×10-22(T-0.47)exp(-9949/T),k的單位為cm3/(molecule·s)。目前,有關該反應速率常數的研究較少,因此此計算結果可為今后進一步研究提供參考。
3 結 論
(1)采用從頭算方法和基組優化反應路徑中各駐點的幾何結構并計算前線軌道能和振動頻率,采用極化連續介質模型研究了反應的溶劑化效應,得到標準態和溶劑化效應下的反應勢能面,對比發現在高溫下反應更傾向于生成穩定的異丙醇和水,其過渡態能壘為82.92kJ/mol。
(2)研究了pH值變化對水解反應的影響,發現pH值的變化對駐點能量以及反應路徑順序影響不大。利用TST、CVT、CVT/ZCT和CVT/SCT方法得到SN2反應速率常數k的Arrhenius表達式為:k=1.18×10-23exp(-7357/T),計算出的反應速率常數相對較小,說明反應速率可能較慢。
致謝:感謝華南師范大學提供Gaussain 09和Polyrate 2015程序包;同時感謝王朝陽老師在研究過程中給予的指導和幫助。
