固相萃取-氣相色譜法測定茶葉中氟環唑
2018-12-03 08:20:02李崢嶸黃曉東
安徽工程大學學報 2018年4期
李崢嶸,黃曉東
(安徽工程大學 生物與化學工程學院,安徽 蕪湖 241000)
氟環唑(epoxiconazole),分子式C17H13ClFN3O,是德國巴斯夫公司研發的內吸型、長效、低毒三唑類含氟殺菌劑,在預防和防治一系列禾谷類作物病害方面取得了良好效果[1-2]. 近年來隨著三唑類殺菌劑的使用和亂用,對生態環境造成極大危害[3-4].2015年,氟環唑被歐盟劃分為“(1類、2類)生殖毒性”和“(2類)致癌”的農藥[5].歐盟規定了氟環唑在茶葉上的MRLS(最大殘留量)值為0.05 mg/kg.目前我國在此方面無明確規定,可能會導致我國茶葉出口在歐盟受阻,故研究氟環唑殘留量檢測方法具有現實經濟意義.
目前對氟環唑殘留量的檢測大多數集中在蔬菜和谷物中,檢測方法有UPLC-MS/MS[6-9],GC-MS/MS[10-11]等.茶葉中的氟環唑殘留分析凈化多用中性氧化鋁[12]、GCB柱(石墨化炭黑)[13]、TPT混合填料柱[14-15]、GPC(凝膠滲透色譜)[16]以及QuEChERs(分散固相萃取)[17]等凈化方法,以上方法均可靠,但步驟較為繁瑣.實驗采用Florlisil柱-氣相色譜法檢測茶葉中氟環唑殘留量,簡化了前處理步驟,提高了實驗精確性和穩定性,是有效監督控制氟環唑殘留量的檢測方法.
1 材料與方法
1.1 材料與試劑
綠茶,產自蕪湖;乙腈、丙酮、正己烷、甲醇、氯化鈉(色譜純或分析純);C18萃取柱;NH2萃取柱;Florlisil柱;氟環唑標樣(濃度≥99%,德國Dr.Ehrenstorfer);超純水.
1.2 儀器與設備
GC-2010氣相色譜儀(配有ECD)(日本島津公司);VX-200渦旋混合器(美國Labnet);旋轉蒸發濃縮儀(上海弘懿儀器設備有限公司);TGL-16G離心機(上海安亭儀器廠).
1.3 實驗方法
(1)氣相色譜條件.色譜柱:DB-17柱(30 m×0.32 mm×0.25 μm);溫度:INJ為250 ℃ ;DET為300 ℃;COL為180 ℃(1 min),8 ℃/min ,250 ℃(10 min);載氣 :高純氮氣 ,柱流量 2.0 mL/min;進樣方式:不分流,進樣量1 μL.
(2)標準曲線的繪制.準確稱取10 mg氟環唑標準品,用正己烷溶解配制成1.0 mg/mL的氟環唑標準儲備母液(不用時置于-18 ℃以下冰箱避光儲存)和0.01 mg/mL的氟環唑儲備液(不用時置于0 ℃~4 ℃冰箱避光儲存).用正己烷稀釋成1.0 μg/mL、0.2 μg/mL、0.1 μg/mL、0.06 μg/mL、0.02 μg/mL的氟環唑標準溶液,在選定色譜條件下進行GC-ECD檢測,并計算氟環唑標準曲線線性關系方程.
(3)樣品前處理.用粉碎機將茶葉粉碎成末,過100目篩.稱取1.0 g茶葉粉樣品于50 mL加蓋離心管中,加入18 mL乙腈和0.4 g NaCl,渦旋混勻,超聲輔助提取12 min,8 000 r/min離心10 min,轉移上清液至雞心瓶中,真空旋轉濃縮近干,加入5 mL正己烷-丙酮(7+3)超聲溶解,上樣至Florlisil柱+GCB混合柱,接出流出液,再每次用2 mL正己烷-丙酮(7+3)3次清洗雞心瓶,并將洗滌液加入柱中,繼續加入20 mL正己烷-丙酮(7+3)洗脫,旋轉濃縮近干,其后加入1 mL甲醇-水(9+1)超聲溶解,渦旋混勻,過0.22 μm濾膜轉移至進樣瓶,供GC-ECD分析.
(4)固相萃取條件選擇.
①提取劑的選擇.準確稱取4組相等的茶葉樣品,同時添加質量濃度為0.05 mg/kg的氟環唑標準液,分別用正己烷、乙腈、丙酮和甲醇作為提取劑,在其他實驗條件保持不變的情況下,按照上述步驟進行樣品前處理,供GC-ECD分析,計算回收率,確定提取劑的種類.
②提取劑體積的選擇.準確稱取5組相等的茶葉樣品,同時添加質量濃度為0.05 mg/kg的氟環唑標準液,只改變提取劑的體積,分別用10 mL、15 mL、20 mL、25 mL和30 mL選定的提取劑提取,計算回收率,確定提取劑的體積.
③NaCl加入量的選擇.準確稱取5組相等的茶葉樣品,同時添加質量濃度為0.05 mg/kg的氟環唑標準液,只改變NaCl固體加入量,NaCl加入量分別為0.0 g、0.4 g、0.8 g、1.2 g、1.6 g,計算回收率,確定NaCl加入量.
④超聲處理時間的選擇.為了使凈化效果達到最佳,實驗采用超聲輔助的方法,超聲時間分別設定為0 min、5 min、10 min、15 min和20 min,其他條件不變,計算回收率,確定超聲處理時間.
⑤洗脫液比例的優化.準確稱取4組相等的茶葉樣品,同時添加質量濃度為0.05 mg/kg的氟環唑標準液,只改變洗脫液正己烷、丙酮的體積比,體積比分別為6∶4、7∶3、8∶2、9∶1,計算回收率,確定洗脫液比例.
⑥固相萃取柱選擇.為了使凈化效果達到最佳,分別選用C18,NH2和Florlisil 3種固相萃取柱,氟環唑標準液質量濃度為0.05 mg/kg,其他條件不變,計算回收率,確定最佳萃取柱.
⑦方法的重復性實驗.在選定的最佳實驗條件下對加標水平為0.05 mg/kg的茶葉樣品進行處理及測定,連續5天每天測一次,計算其精密度.
⑧方法的回收率實驗.取3份等量的茶葉樣品,選擇3種不同質量濃度的氟環唑標準液進行加標,添加水平分別為0.02 mg/kg、0.05 mg/kg、0.1 mg/kg,采用最佳的實驗條件進行分析,連續測定5次,取平均值,計算回收率和變異系數.
⑨結果計算公式.氟環唑質量含量c1(ug/g)按下式計算
式中,c1為樣品中氟環唑含量(μg/g);c為儀器檢測的質量濃度(mg/L);V為最終定容體積(mL);m為樣品質量(g).
2 結果與討論
2.1 提取劑的選擇
氟環唑在不同提取劑中的溶解度是不一樣的,在滿足實驗要求的前提下,不同的提取劑對氟環唑的回收率有一定的影響.提取劑對氟環唑回收率的影響如圖1所示.由圖1可知,乙腈和丙酮作為提取劑,回收率較高,實驗過程中發現丙酮萃取液中含有大量的色素,導致后續的凈化過程存在一定的困難,故選擇乙腈作為提取劑.
2.2 提取劑體積的選擇
選定乙腈作為提取劑,改變乙腈的體積,考察乙腈體積對氟環唑回收率的影響, 結果如圖2所示.由圖2可知,乙腈體積為20 mL時回收率最高;乙腈體積小于20 mL時,提取的不夠充分;乙腈體積大于20 mL時,導致后續濃縮過程的氟環唑的損失,故選擇最優提取劑體積20 mL.
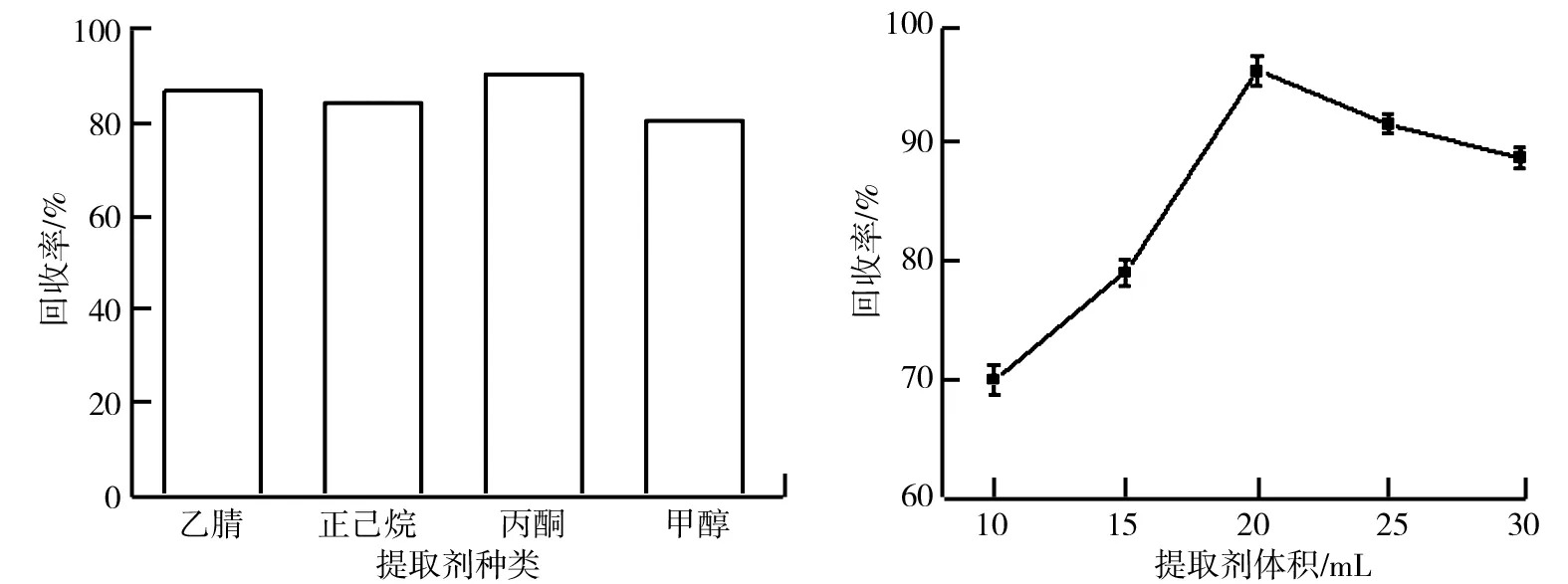
圖1 提取劑對氟環唑回收率的影響 圖2 提取劑體積對氟環唑回收率的影響
2.3 NaCl加入量的選擇
在樣品溶液中加入NaCl會降低待測物在水相中的溶解度、制約萃取劑在水中的溶解性,從而有利于對待測組分回收.NaCl質量對氟環唑回收率的影響如圖3所示.由圖3可知,在NaCl的用量逐漸增加時氟環唑的回收率先增加后降低,在NaCl的用量為0~0.8 g時,回收率呈線性增加,NaCl加到0.8 g時成為轉折點,隨著NaCl質量濃度增高導致萃取效果變差,回收率降低.因此選擇0.8 g作為NaCl的最佳用量.
2.4 超聲處理時間的選擇
樣品的前處理過程中,超聲輔助萃取可以提高萃取效率.實驗具體考察了不同的超聲處理時間對氟環唑回收率的影響,結果如圖4所示.由圖4可知,超聲處理時間由0 min到15 min時,萃取效果在提高,繼續超聲處理,其促進作用達到了飽和,可能是因為氟環唑見光易分解的特性,長時間暴露在日光中,有部分損失掉了,使得氟環唑的回收率開始下降,綜合考慮超聲時間為15 min時最佳.
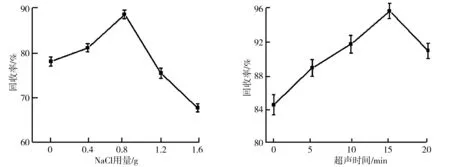
圖3 NaCl質量對氟環唑回收率的影響 圖4 超聲時間對氟環唑回收率的影響
2.5 洗脫液比例的優化
由于樣品的基質較雜,合適的洗脫液可使目標物盡可能從柱中洗脫下來,同時減少雜質被洗脫下來,對比研究了不同體積比洗脫液的洗脫效果,結果如圖5所示.由圖5可知,氟環唑的回收率隨著洗脫液體積比的減小先增高后降低,在體積比為7:3時回收率較高,可能是由于洗脫液的極性偏大或偏小,不利于目標物的洗脫凈化,導致氟環唑的回收率較低.
2.6 固相萃取柱選擇
樣品中的基質成分復雜多樣,必須經過凈化處理,不同的萃取柱性能、效果不同,合適的萃取柱可降低檢測器的雜峰干擾,提高回收率.為了最大限度地排除雜質離子的干擾,對比研究了不同種類的固相萃取柱的凈化效果,結果如圖6所示.由圖6可知,Florlisil柱的凈化效果最好.
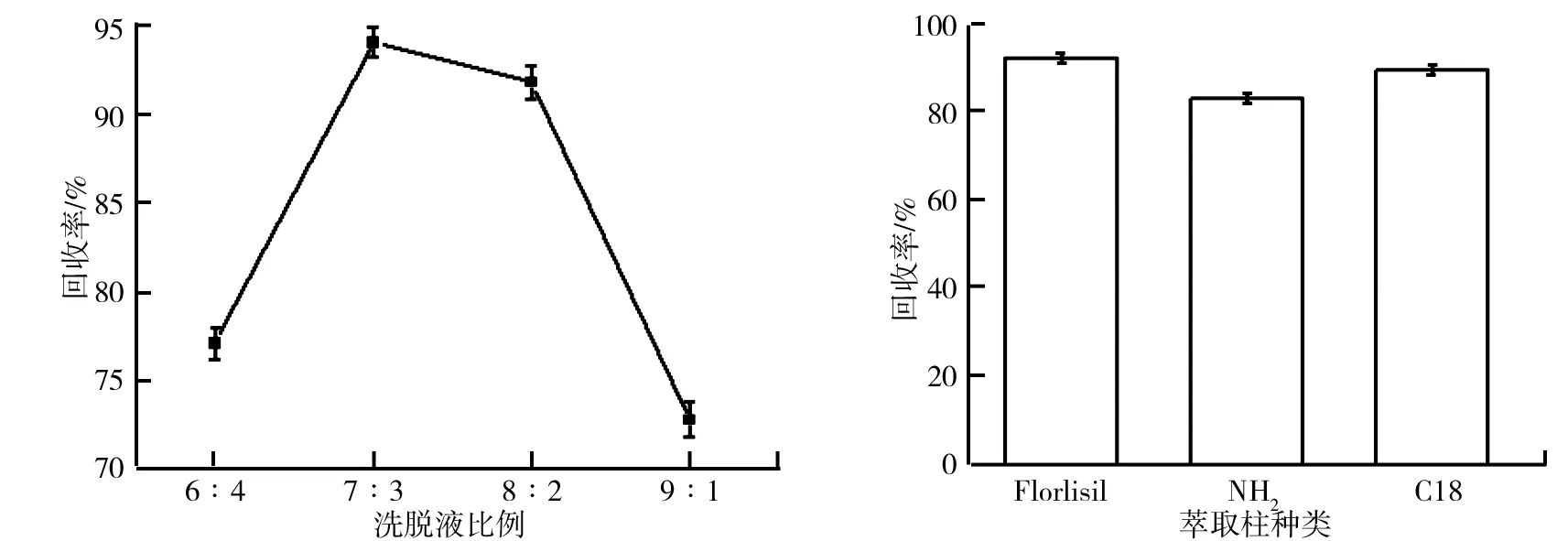
圖5 洗脫液比例對氟環唑回收率的影響 圖6 萃取柱種類對氟環唑回收率的影響
2.7 標準曲線的繪制
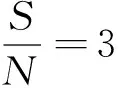

圖7 氟環唑標準曲線 圖8 氟環唑標準溶液氣相色譜圖(1.275 μg/mL)
2.8 響應面分析優化氟環唑的提取條件
(1)響應面試驗因素水平的選擇.實驗根據以上單因素實驗結果,選超聲時間、氯化鈉用量和提取劑體積為主要考察因素,以氟環唑的回收率為指標,設計3因素3水平響應面試驗,選用Design-Expert 8.0軟件中Box-Behnken作為響應面模型.實驗的因素及水平如表1所示.

表1 響應面試驗
(2)響應面實驗設計及結果分析.以A(超聲時間)、B(NaCl用量)、C(提取劑體積)為自變量,氟環唑的回收率為Y(響應值),以此進行17組分析試驗,其中用以估算誤差中心試驗有5個.BBD的試驗結果如表2所示.根據表2中試驗的結果應用Design-Expert 8.0軟件進行分析,3個自變量模型的方差分析結果如表3所示.由表3可知,模型的F=16.77,P=0.000 6<0.001,表明實驗的二次模型是極顯著的,在統計學上有意義.實驗失擬項P=0.131 4>0.05,對模型有利,無失擬因素,數據中無異常點出現,因此可用于預測各因素與氟環唑回收率的關系.
由Design-Expert 8.0軟件得出各個因素的效應圖如圖9、圖10、圖11所示.由圖9、圖10、圖11可知,圖形的表面有明顯彎曲,可得各個因素對響應值Y是非線性的,A、B、C三個因素水平存在極值點.
(3)響應面回歸模型的建立.結合Design-Expert軟件計算,可得該響應面模型的回歸方程如下:
R=-281.546 25+22.601 25A+205.528 12B+12.283 5C-3.275AB-2.7E-003AC-5.076 25BC-0.629 8A2-42.125B2-0.214 9C2
(4)各因素水平的優化.根據效應圖繪制回歸方程的3D響應面圖,回歸方程存在極值點.為了確證其最佳值,對方程取一階偏導,整理可得:
22.601 25-3.275B-2.7E-003C-1.259 6A=0
205.528 12-3.275A-5.076 25C-48.25B=0
12.283 5-2.7E-003A-5.076 25B-0.425 8C=0
解方程組得A=13.02,B=0.75,C=19.65,代入響應面方程得到氟環唑的最大響應值是87.31.即在超聲時間13 min,NaCl用量0.7 g,提取劑乙腈20 mL時,氟環唑的回收率高達87.31%.
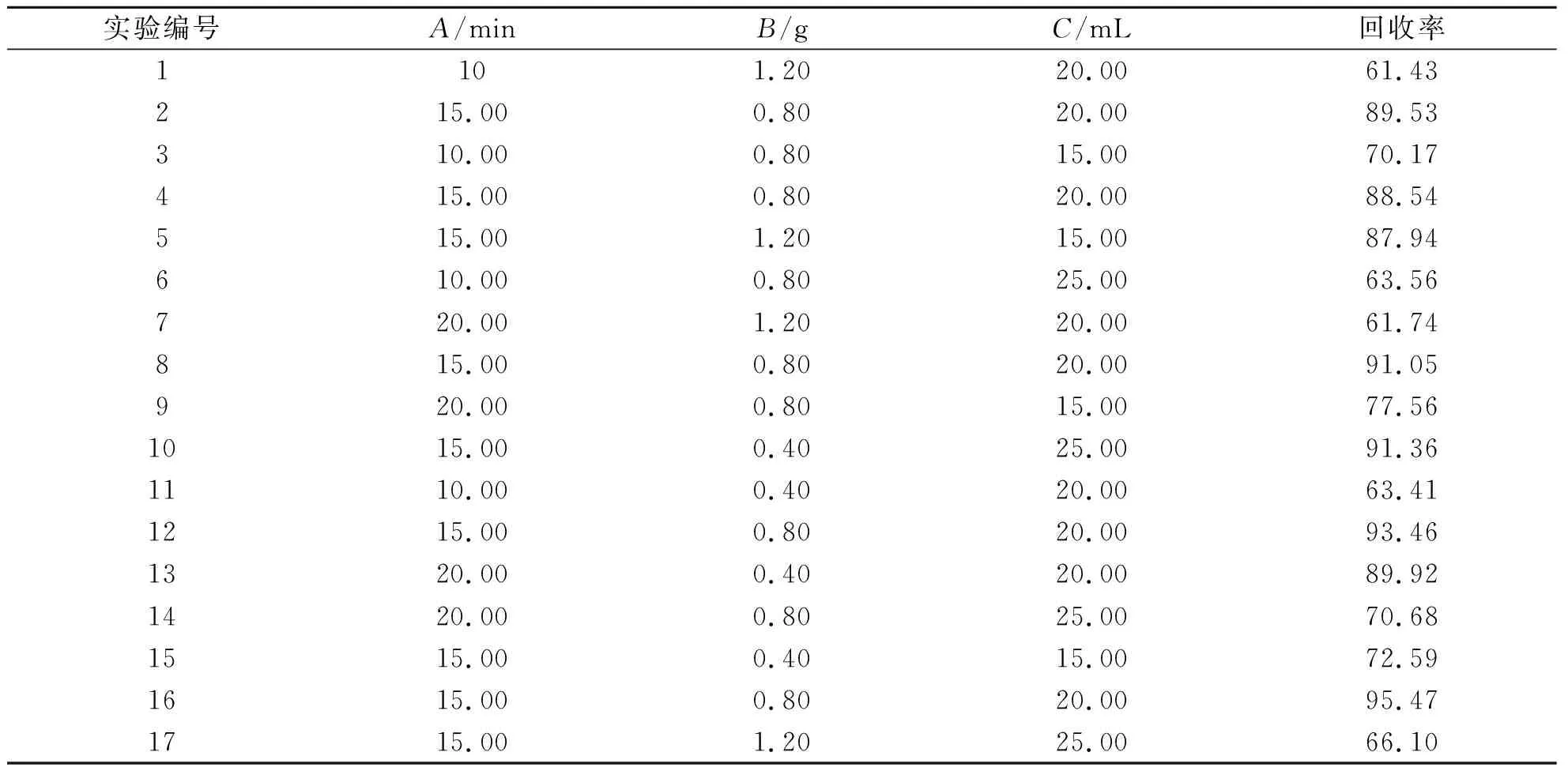
表2 BBD的試驗結果
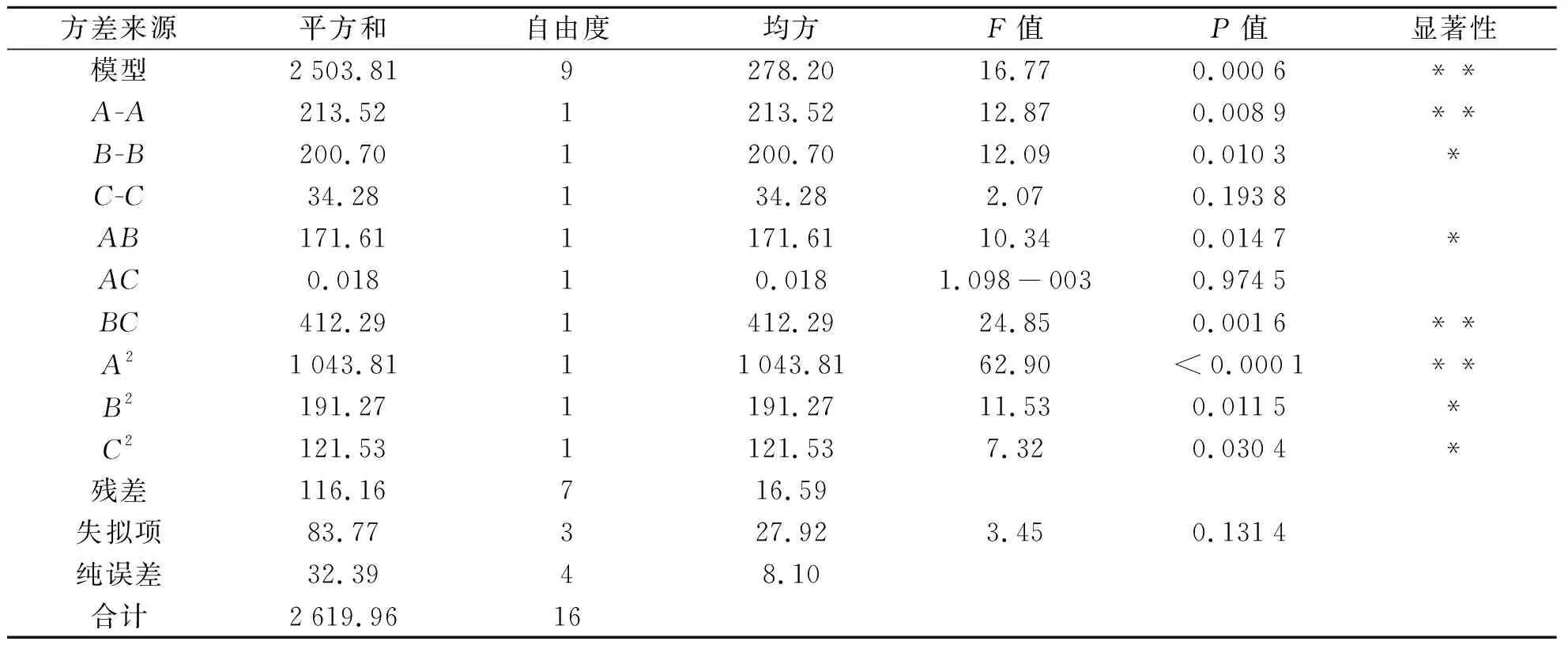
表3 響應面試驗的方差分析結果
P<0.05時,影響顯著;P<0.01時,影響極顯著,分別用*和**表示
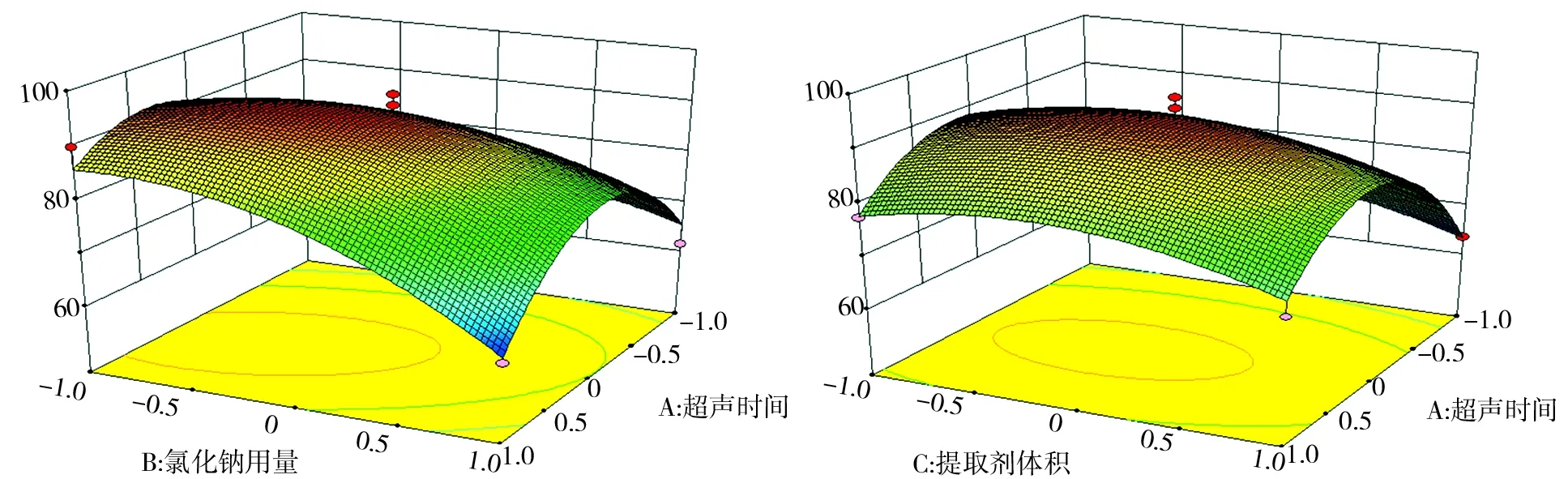
圖9 A和B對氟環唑回收率的影響 圖10 A和C對氟環唑回收率的影響
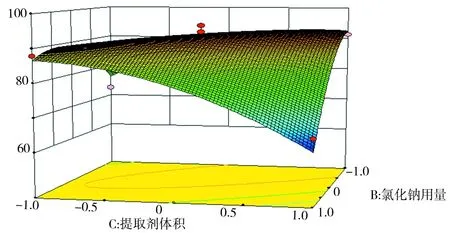
圖11 B和C對氟環唑回收率的影響
2.9 方法的重復性實驗
準確稱取茶葉樣品,按上述方法進行測定,結果如表4所示.由表4可知,樣品中氟環唑質量濃度的平均值是0.044 7 mg/kg,RSD為3.55%,方法重復性和精密度較好.
表4方法精密度實驗
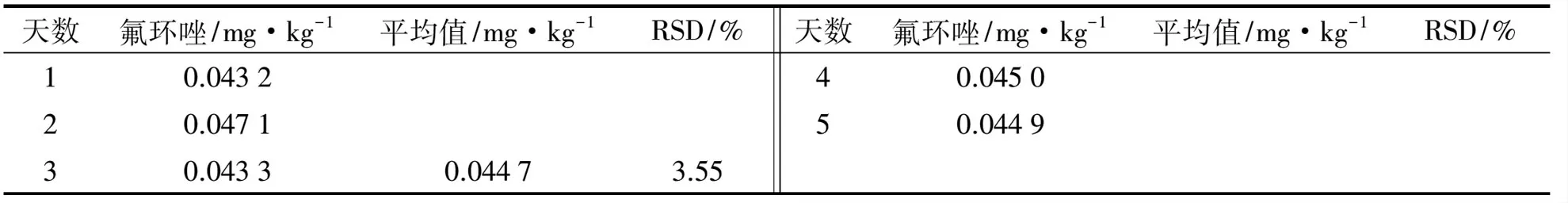
天數氟環唑/mg·kg-1平均值/mg·kg-1RSD/%10.043220.047130.04330.04473.55
2.10 方法的回收率實驗
分別稱取3份茶葉樣品,在樣品中分別加入0.02 mg/kg、0.05 mg/kg、0.1 mg/kg 3種質量濃度的氟環唑標準液,以最佳條件進行實驗處理,結果如表5所示.由表5可知,氟環唑在茶葉中的添加回收率范圍為86.38%~92.06%,平均回收率為89.32%,且變異系數范圍為3.06%~5.64%,可見方法的回收率較好.茶葉粉中氟環唑的添加回收色譜圖(加標水平為0.127 5 mg/kg)如圖12所示.
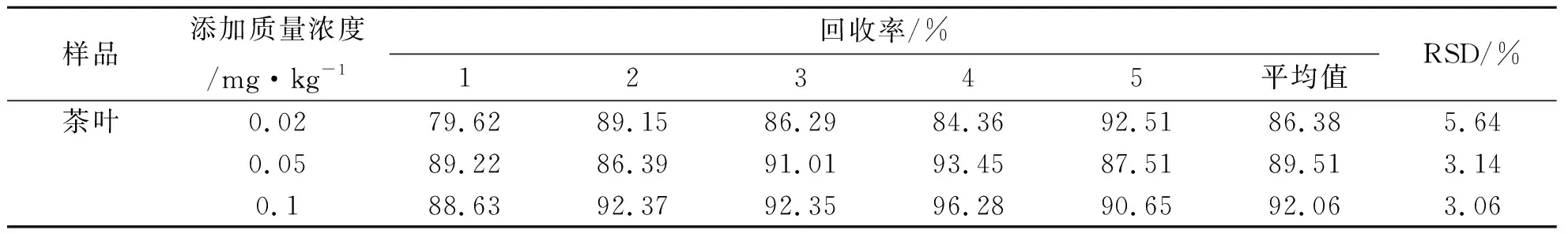
表5 方法的回收率實驗
圖12 氟環唑在茶葉中的添加回收色譜圖
3 結論
SPE-GC測定茶葉中的氟環唑殘留量的實驗中,樣品凈化步驟采用Florlisil柱,應用Design-Expert 8.0優化樣品預處理方法,得到最佳條件下回收率為87.31%,加標回收率在86.38%~92.06%之間,重復性實驗RSD為3.55%,最低檢出限為0.007 μg/mL.實驗操作簡單、快速、穩定,方法的精密度、重現性、靈敏度、回收率、線性關系良好,可用于大量樣品的檢測.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
兒童故事畫報(2019年5期)2019-05-26 14:26:14
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
