乳品中的酵母活菌數PMAqPCR檢測方法的建立及初步應用
2018-12-07 07:02:16巖松
食品工業科技 2018年22期
, , , , ,,,,*, ,,,巖松, ,
(1.工商學院糧油食品深加工吉林省高校重點實驗室,吉林長春 130507;2.人獸共患病研究教育部重點實驗室,吉林大學人獸共患病研究所,吉林大學動物醫學學院,吉林長春 130062;3.吉林廣澤乳業有限公司,吉林長春 130102)
在乳品行業的發展過程中,微生物發揮了極其重要的作用,尤其在發酵乳中含有大量的益生菌,有利于乳品的消化吸收[1-2],但微生物污染也是影響乳品質量的一個重要因素,乳品常見的污染微生物為細菌、酵母菌與霉菌,微生物超標會導致乳品變質、腐敗、脹袋[3],而酵母菌污染是導致乳品變質脹袋的主要原因[4]。頻發的乳品安全事件不僅影響了消費者健康,還使外資乳品企業有機可乘占領國內市場,嚴重制約了我國乳品行業的發展[5]。為了確保乳品質量合格,國標中明確規定了乳品中微生物限量標準[4]。目前,乳品加工企業最常用微生物檢測方法有微生物培養法(平板計數法)、ATP 生物發光法和儀器法等,但各技術均有不足。如平板計數法雖操作簡便,成本低廉,但耗時太長,易漏檢 VBNC 狀態微生物[6];流式細胞儀法。雖準確快速,但儀器昂貴,檢測成本高[4-7]。近年來,EMA/PMA 結合 qPCR 檢測復雜環境樣品中活菌的方法得到了一定的發展[8]。有研究利用PMA-qPCR檢測實現了區分檢測排放的生活廢水中的金黃色葡萄球菌活菌數[9]。另外,有研究利用PMA-qPCR檢測環境樣品中的活鉤蟲卵的數量[10]。本研究目的是開發一種靈敏的PMA-qPCR法,可有效區分死菌與活菌,定量檢測存在于培養基中的活的以及活的但不可培養的(VBNC)酵母菌[11]。
疊氮溴化丙錠(propidiummonoazide,PMA)是可以進入細胞膜損傷的死亡酵母菌中,與DNA雙鏈發生共價交聯反應的染料,結合后能抑制死菌DNA的PCR擴增,而活菌完整的細胞膜可阻止PMA進入細胞內[7]。優化PMA處理條件,建立乳品中酵母菌的PMA-qPCR活菌檢測方法,再結合熒光定量PCR確定國標中酵母菌總數最大限量值所對應的Ct值范圍[7],PMA-qPCR法將實時熒光定量PCR的快速和高度特異性與PMA對細胞膜損傷酵母菌的選擇透過性結合起來,降低對酵母活菌總數檢測的干擾和假陽性。同時與傳統國標平板計數法相比[7],更加快速高效。從污染菌很少的樣品中快速富集,以集菌方法代替增菌培養,結合實時熒光檢測,能夠縮短檢驗時間,提高檢測效率[12]。該方法用于判斷乳品中的活菌總數是否超標,為乳品的生產加工提供有效的活菌監控方法,以確保乳品的質量與安全。針對以上問題,本實驗從廣澤乳業的乳品樣品中分離污染的酵母菌,旨在建立能區別死菌和活菌的快速PMA-qPCR檢測方法。
1 材料與方法
1.1 材料與儀器
乳品 均來自廣澤乳業商品巴氏乳;疊氮溴化丙錠(PMA) Biotium公司;2×PCR預混酶Easy Taq Master Mix 北京全式金公司;2×SYBR熒光定量PCR預混酶Fast Start Universal SYBR Green Master(Rox) Roche公司;酵母菌基因組DNA提取試劑盒 天根生物技術有限公司;膠回收試劑盒 Takara公司;卡那霉素 北京鼎國昌盛生物技術有限責任公司;菌株:馬克思克魯維酵母菌、庫德里阿茲威畢赤酵母菌、熱帶假絲酵母菌 由本實驗室保藏。
650 W鎢絲燈 OSRAM公司;ABI7500熒光定量PCR儀 美國應用生物系統公司;紫外凝膠成像系統 UVP公司。
1.2 實驗方法
1.2.1 酵母菌活菌懸液和死菌懸液的制備及國標定量檢測 從YPD平板挑取酵母菌單菌落,接種于5 mL的YPD液體培養基中[7],分別加入5 μL卡那霉素和氨芐青霉素,220 r/min振搖,28 ℃恒溫培養23~28 h,至OD600為0.5。
酵母菌定量檢測在國標GB4789.2-2016基礎上進行改良,即在平皿中加入1 mL稀釋后的不同梯度稀釋的菌液和11 μL卡那霉素后,倒入固體培養基,輕微晃動平板至混合均勻,靜置30 min至平板凝固,28 ℃培養48~72 h。此后操作均按照此改良國標法對三種酵母菌進行平板計數。
采用改良后平板法計算活菌懸液濃度。將活菌懸液8000 r/min離心10 min,按照計數結果,將酵母菌以無菌生理鹽水重懸至密度約1×103CFU/mL后,沸水浴10 min,制備死菌懸液,冷卻到室溫備用。
1.2.2 PMA最佳工作濃度的選擇 設置PMA 6個濃度梯度1、3、5、7、9 μg/mL分別加入1 mL數量級為1×103CFU/mL的死菌懸液中[7],渦旋振蕩器振蕩混勻避光孵育10 min后,置于冰上用650 W燈泡充分曝光10 min,煮沸10 min,以處理后的菌液為模板進行qPCR,選取沒有擴增曲線的最小抑制濃度[4],確定PMA的最佳工作濃度。
1.2.3 PMA-qPCR方法的建立
1.2.3.1 熒光定量通用引物設計 Primer Premier 5.0設計酵母26SrDNA熒光定量通用引物,Y3:5′-CGAGTTGTTTGGGAATGC-3′,Y4:5′-ACTTGTTC GCTATCGGTCTC-3′,引物由吉林庫美生物技術有限公司合成,擴增產物長度90 bp。提取三種酵母菌DNA為模板進行qPCR,獲得擴增目的條帶,進行電泳。反應條件為:95 ℃預變性30 s,95 ℃變性5 s,57 ℃退火40 s,40個循環。在每一循環的退火階段收集熒光,實時檢測并記錄熒光信號強度,得到熔解曲線。
1.2.3.2 pMD-18T-26S標準質粒的構建 按照基因組提取試劑盒說明書提取酵母菌DNA,以此為模板進行PCR,產物進行1%瓊脂糖凝膠電泳,用膠回收試劑盒對PCR產物進行膠回收。按照pMD-18T Vector使用說明書,16 ℃水浴反應連接過夜[13]。將連接產物轉化進E.coliDH5α(DE3)感受態細胞后,取50 μL在含有氨芐青霉素抗性的瓊脂培養基上反復涂布至均勻,37 ℃倒置培養12 h。挑取單個圓潤明亮的白色菌落,于5 mL LB培養基中,37 ℃恒溫180 r/min振搖12 h[13]。菌液PCR法鑒定正確的菌液進行測序,確認載體中插入片段的長度大小,同時送往生物科技公司進行克隆測序,進行序列比對。
1.2.3.3 熒光定量標準曲線的建立 按照質粒提取試劑盒的說明提取質粒后,檢測濃度后計算拷貝數,用無菌生理鹽水梯度稀釋至1×102~1×109。以此為模板進行qPCR檢測,每個數量級三個重復孔,取平均值,以pMD-18T-26S拷貝數對數與Ct值為坐標軸,建立標準曲線[13]。
1.2.3.4 PMA對qPCR的干擾性驗證 對經過PMA處理和未處理的1×103CFU/mL的酵母菌分別進行qPCR,對比兩組樣品的溶解曲線與擴增曲線。
1.2.4 乳品中酵母菌PMA-qPCR活菌檢測方法評價
1.2.4.1 靈敏度檢測 取溶解曲線呈單峰且擴增曲線良好的酵母菌最小檢測濃度為酵母最低檢測濃度,確定 PMA-qPCR法對酵母菌個數的最低檢測限。
1.2.4.2 重復性檢測 按照1.2.3.3中的qPCR體系進行組間穩定性測試,據前述PCR反應條件進行熒光PCR檢測。在不同的時間內分別做3次,計算擴增循環數(Ct值)的標準差與CV值[14]。
1.2.4.3 PMA-qPCR樣本的前處理及加標回收實驗 量取50 mL乳品于離心管中,經8000 r/min高速離心15 min[4],棄掉脂肪,重復兩次,將處理過的乳品稀釋液于離心管中分裝,65 ℃放置2 h滅菌后[15],將乳品稀釋液密封4 ℃保存。取處理好的乳品稀釋液各1 mL于40個離心管中,隨機添加酵母活菌與添加熱致死菌[7]模擬加標乳品,編號1~40。加標乳品經PMA處理后進行qPCR,每個樣本三個重復孔。將PMA-qPCR結果與改良國標計數對比。
1.2.5 實際乳品樣品酵母總數PMA-qPCR檢測 對從廣澤乳業采集的不同批次乳品樣本,采用擴大檢測體積10倍并離心的方法檢測濃度,并隨機取一部分置于37 ℃增菌培養[16],共80個樣品,編號1~80,對PMA-qPCR檢測結果進行分析。
1.3 數據統計分析
使用Excel對原始數據進行初步分析,使用SPSS 22.0計算標準偏差σ來量度每組數據分布的分散程度[17]。
2 結果與分析
2.1 目的條帶的擴增
用Y3和Y4引物擴增的三株酵母菌的DNA片段大小在90~100 bp處均有條帶,與預期結果一致,陰性對照無條帶(圖1)。回收產物經測序檢驗結果正確,證明為目的片段。
2.2 PMA最佳工作濃度的選擇
根據qPCR結果,當PMA濃度為1、3、5 μg/mL時,熔解曲線正常且均有擴增曲線(圖2),說明死菌DNA未被完全結合,擴增時仍能造成假陽性;當PMA濃度為7、9 μg/mL時均沒有擴增曲線(圖3),說明死菌DNA被完全結合,DNA擴增被抑制,由此可得,PMA濃度為7 μg/mL時,是可以完全結合1×103CFU的酵母菌的最低抑制濃度,取7 μg/mL為最佳PMA工作濃度。

圖2 熔解曲線Fig.2 Melt curve

圖3 擴增曲線Fig.3 Amplification curve注:1:1 μg/mL;2:3 μg/mL;3:5 μg/mL。
2.3 PMA對qPCR的干擾性驗證
對經過PMA處理和未處理的1×103CFU/mL的酵母菌分別進行qPCR,擴增曲線均正常(圖4);PMA處理組Ct值為36.64±0.52,略大于無PMA組的Ct值35.99±0.53,但二者Ct值基本一致,證明PMA對qPCR的干擾極小。
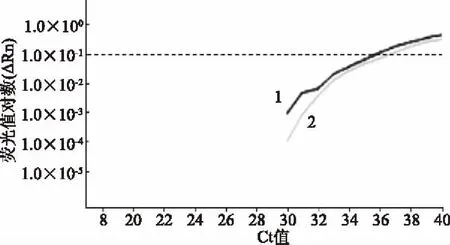
圖4 擴增曲線Fig.4 Amplification plot注:1:無PMA組;2:PMA處理組。
2.4 PMA-qPCR標準曲線的建立
檢測Ct值與重組質粒pMD-18T-26S拷貝數對數線性關系較好,熔解曲線呈單峰,特異性良好(圖5)。標準曲線為y=-4.7564x+50.94,R2為0.9952,線性關系良好,檢測范圍為:1×103~1×109CFU/mL。
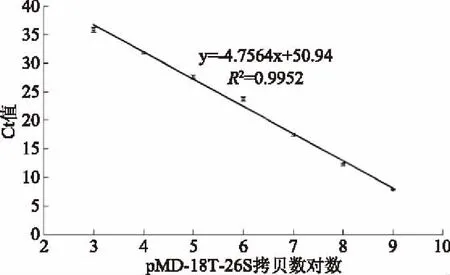
圖5 標準曲線Fig.5 The standard curve
2.5 樣本處理分析
PMA-qPCR檢測濃縮樣品與未濃縮樣品酵母菌數量,濃縮組的Ct值略高于未濃縮組的Ct值,分析原因是濃縮操作過程中有少量酵母菌損失所致;隨著酵母菌濃度數量級的增大,兩組樣品的變異系數逐漸減小,各組變異系數差異均小于10%(表1)。經濃縮策略處理,該方法靈敏度提高,方法實際檢測限最低可達1×102CFU/mL酵母菌。
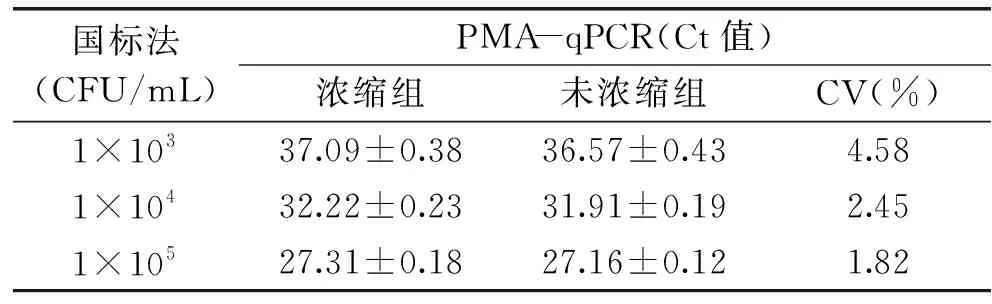
表1 濃縮與未濃縮樣品結果比對Table 1 Results of concentrated and unconcentrated samples
2.6 乳品PMA-qPCR檢測方法靈敏度分析
在酵母濃度為1×103CFU/mL時,溶解曲線呈單峰,且特異性良好,最低檢測范圍為1×103CFU/mL,檢測范圍為對應1×103~1×109CFU/mL,Ct值范圍為8.13±0.11~36.67±0.43。
2.7 乳品PMA-qPCR檢測方法重復性檢驗
對1×103~1×105數量級的酵母菌分別進行三次qPCR檢測,對三次結果進行分析,計算平均值、標準差和變異系數,變異系數均小于10%,證明該方法重復性良好(表2)。

表2 重復性檢驗Table 2 Detection of duplicability
2.8 加標回收結果
乳品中酵母菌濃度分別在1×103~1×104CFU/mL范圍內和在1×104~1×105CFU/mL范圍內時,國標法與PMA-qPCR法檢測結果重合率為分別為93.75%和95.00%(表3)。分析可得,當乳品中酵母總數處于1×104~1×105的數量級時相比較于1×103~1×104的數量級,PMA-qPCR法與改良國標法檢測結果越接近,結果更加準確可信。

表3 乳品加標回收結果Table 3 Determination for dairy products
2.9 實際樣品酵母菌數量檢測
檢測市售樣品和增菌培養樣品共80份,其中檢測市售廣澤乳業乳品20份中7份樣品未檢出酵母菌,且13份檢出樣品酵母菌數量均在國標規定范圍內;檢測增菌樣品60份,酵母濃度多處于1×102~1×104CFU/mL,增菌培養后的樣品濃縮后PMA-qPCR法檢測結果與國標法重合率約為93.33%以上(表4),實際樣本檢測總耗時6~7 h。

表4 乳品樣品檢測結果Table 4 Test results of milk samples
3 討論
3.1 運用PMA去除熱損傷酵母菌的意義
自Nocker等驗證了PMA可以替代EMA從樣本中準確計數活細胞后,PMA與分子生物學方法結合技術已被廣泛應用于環境和食品等多個領域的微生物檢測研究[4,18]。如果只采用qPCR法,死亡酵母菌的DNA穩定存在,同樣被qPCR檢測[19],本實驗應用PMA結合死菌的DNA而使其不能進行qPCR擴增,可避免假陽性情況的出現。本方法實現了對復雜乳品樣本中損傷酵母菌DNA的有效去除,降低對酵母活菌總數檢測的干擾,降低假陽性檢測結果出現的可能性[19]。
首先需要確定酵母菌濃度在不同數量級時,所需的PMA最低抑制濃度。有研究表明,當酵母菌濃度為1×107CFU/mL時,所需PMA最低抑制濃度為15 μg/mL[4],本方法為了盡可能達到酵母菌數量國標規定檢測限1×102CFU/mL,選取酵母菌濃度1×103CFU/mL時,進行PMA工作濃度的優化,確定PMA最低抑制濃度為7 μg/mL,作為本方法的最佳工作條件。但也有研究表明,由于PMA會輕微抑制PCR反應,傳統上定量分析PCR采用終點法存在缺陷,而實時技術可以彌補其缺陷,可以實時地在PCR反應擴增中進行檢測,只要少量的DNA模板,熒光信號經過連續的分析,就會擴增得到曲線[20]。
3.2 酵母DNA提取體系的優化
雖然根據郭冬琴等[21]的研究結果表明,熒光定量檢測乳制品中的酵母菌不受食品基質的影響。但提取高質量的DNA作為模板是進行分子鑒定的基礎,所以讓乳品中的酵母菌DNA充分暴露是很關鍵的一步。常用的方法有石英砂或玻璃珠破碎和超聲破碎酵母菌體[22],本方法采用最原始有效的菌體煮沸破碎法,將PMA處理后的實際樣品煮沸10 min,使DNA充分暴露后再電泳等改良措施提高DNA的提取數量和質量。既不需要消耗太多成本,又可以達到良好的檢測效果。
4 結論
建立了PMA-qPCR定量檢測乳品中酵母活菌總數的技術。確定酵母菌為1×103CFU/mL時,PMA最佳工作濃度為7 μg/mL。將樣品煮沸10 min,使酵母菌DNA充分暴露,通過qPCR使序列充分擴增,得到準確的Ct值36.67±0.43。通過濃縮實際乳品樣本降低對酵母菌總數的最低檢測限,最低檢測限可達到1×102CFU/mL,與國標法重合率高于93.33%。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
軍事文摘·科學少年(2021年1期)2021-02-04 08:03:45
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
閱讀與作文(小學低年級版)(2015年8期)2015-05-30 10:48:04
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
食品工業科技(2014年9期)2014-03-11 18:15:31
