超高效液相色譜串聯質譜法測定食品中柑橘紅2號和蘇丹紅Ⅰ~Ⅳ染料
2018-12-10 01:34:10,,,,,,,
食品工業科技 2018年23期
關鍵詞:效應
,, ,, , ,,
(山東省食品藥品檢驗研究院,山東濟南 250101)
柑橘紅2號和蘇丹紅Ⅰ~Ⅳ 均屬于脂溶性偶氮類染料,主要在石油、機油和其他的一些工業溶劑中作為工業染料使用[1]。因這些工業染料具有著色穩定、色澤艷麗、價格低廉等特點,一些不法商家將其添加到食品中進行染色,以提高其市場競爭力。柑橘紅2號和蘇丹紅染料在化學結構上均具有偶氮骨架,這種結構決定了其具有一定的致癌性[2]。據世界衛生組織的國際癌癥研究機構(IARC)1995年確定,蘇丹紅屬第三類致癌物質;歐盟國家已在1995年禁止其作為食用色素,我國已明確將蘇丹紅列在《食品中可能違法添加的非食用物質和易濫用的食品添加劑名單》中。世界衛生組織認為柑橘紅2號具有致癌危險,對人體的肝臟、腎臟器官具有明顯的毒副作用,也不能作為食品添加劑用于食品中[3]。但近幾年各國仍不斷有食品中添加化工染料的食品安全事件發生。一些不法食品企業把蘇丹紅添加到食品中,曾引發“紅心鴨蛋”、“亨氏辣椒醬”等食品安全事件。而近幾年水果市場也相繼發現“染色橙”食品安全問題,香港食物環境衛生署抽查39個橙類樣品,發現1/3的樣品橙皮中含禁用及可能致癌的柑桔紅2號色素。濫用非法染料給食品安全及消費者健康帶來了巨大的隱患。因此,建立快速、準確且能同時檢測柑橘紅2號和蘇丹紅的檢測方法,對食品中非法染料的市場監管具有重要意義。
柑橘紅2號和蘇丹紅染料的測定方法有薄層色譜法、液相色譜法、液相色譜-質譜聯用法、氣相色譜法及酶聯免疫測定法(ELISA)等方法[4-7]。其中以液相色譜法應用最為廣泛,但液相色譜法存在靈敏度較低,特別對于復雜基質,無法滿足痕量檢測的定性定量等問題[5-10]。高效液相色譜-三重四級桿串聯質譜因其高靈敏度、高選擇性及定量定性準確等特性,成為目前此類禁用物質的最佳檢測方法[11-14]。前處理過程方面有直接提取法[15]、氧化鋁柱凈化方法[16]、凝膠滲透色譜(GPC)凈化方法[17]和氨基柱凈化方法[1,3]。其中,直接提取和氧化鋁凈化的方法去除背景干擾能力差,凝膠滲透色譜凈化的方法耗時長,耗溶劑多,易交叉污染。氨基固相萃取柱具有正相和陰離子交換雙重保留作用,被應用于復雜基質中結構相近的化合物的檢測中。目前對辣椒制品等食品中蘇丹紅的研究報道較多,而同時檢測多種基質中柑橘紅2號和蘇丹紅染料的方法鮮有報道[17-20]。
本研究采用乙腈提取,NH2固相萃取柱凈化富集,高效液相色譜-三重四級桿串聯質譜儀檢測,以期建立適用于水果、蜜餞、辣椒制品、火鍋底料及肉制品中柑橘紅2號和蘇丹紅染料同時檢測的方法。并對該方法的靈敏度、選擇性、檢出限進行評價,為食品中痕量水平非法染料的定性、定量檢測提供參考。
1 材料與方法
1.1 材料與儀器
臍橙、蜜餞(山楂卷)、辣椒油、火鍋底料(紅油半固體)及香辣肉灌腸等樣品 均購自濟南當地超市;柑橘紅2號(純度≥90.0%)、蘇丹紅Ⅰ~Ⅳ標準物質(純度≥90.0%) 上海安譜科學儀器有限公司,4 ℃保存;甲醇、乙腈(色譜純) 美國Fisher公司;甲酸、醋酸銨(色譜純) 美國Sigma-Aldrich公司;水 為Milli-Q超純水機制備;氮氣(>99.999%);氨基固相萃取柱(500 mg,3 mL)、色譜柱 Acquity UPLC BEH C18(100 mm×2.1 mm,1.7 μm) 美國Waters公司;有機微孔濾膜(0.22 μm) 上海安譜科學儀器有限公司。
ACQUITYTM超高效液相色譜儀、Xevo TQ-S質譜儀配有電噴霧(ESI)電離源和MasslynxTM色譜工作站 美國Waters公司;Sigma 3-18K高速冷凍離心機 德國Sigma公司;超聲波清洗器 寧波新芝生物科技有限公司;MS3渦旋混合器 IKA公司;N-EVAP-45位氮吹儀 美國Organomation公司;SQP-電子天平 塞多利斯科學儀器有限公司;Milli-Q超純水系統 美國Millipore公司;粉碎機 德國萊馳公司。
1.2 實驗方法
1.2.1 標準儲備溶液的配制 準確稱取各標準物質適量,用乙腈溶解并定容,配制成200 μg/mL標準儲備液,于4 ℃條件下保存,分別吸取各標準儲備液配制成10 μg/mL混合標準工作液,4 ℃條件下保存。
1.2.2 色譜及質譜條件
1.2.2.1 質譜解析及條件的優化 柑橘紅2號同蘇丹紅染料均為含有萘母體骨架的偶氮染料,因此對其較為相似的子離子碎片進行解析,對于此類物質的準確定性具有較好確證作用。在二級質譜全掃描模式(Q1為單離子監測SIM,Q3為全掃描full scan)下獲得該目標物的碎片離子指紋圖譜。二級全掃描模式為定性的強有力手段,但在定量上相比多反應檢測掃描(MRM)靈敏度還是不足,選用MRM掃描模式,確定定性離子對及定量離子對,并優化裂解電壓、碰撞能量等參數。
1.2.2.2 色譜條件的優化 在優化的質譜條件下,考察不同流動相對目標物質譜信號的影響。選用BEH C18色譜柱,分別以甲醇-水、乙腈-水、乙腈-10 mmol/L乙酸銨溶液和乙腈-0.1%甲酸水溶液進行實驗。
1.2.2.3 色譜條件 色譜柱:BEH C18(100 mm×2.1 mm,1.7 μm);流動相:乙腈(A)和0.1%甲酸-水溶液(B),梯度洗脫程序(A):0~2.0 min:10%→50%;2.0~3.0 min:50%→95%;3.0~7.0 min:95%保持4 min;7.0~7.1 min:95%→10%;7.1~9.0 min:10%保持。流速:0.3 mL/min;柱溫:40 ℃;進樣體積:2 μL。
1.2.2.4 質譜條件 離子源:電噴霧離子源(ESI);離子化模式:正離子模式(ESI+);毛細管電壓:3.0 kV;錐孔電壓:25 V;脫溶劑氣溫度500 ℃;離子源溫度:150 ℃;脫溶劑氣流速:850 L/h;錐孔氣流速:150 L/h;碰撞氣流速:0.12 mL/min;掃描方式:多反應監測(MRM);5種非法染料的定性離子定量離子對及質譜參數見表1。
1.2.3 前處理條件的優化
1.2.3.1 樣品提取劑的選擇 考慮到5種偶氮染料的親脂性特點和各種食品等基質較復雜等原因,對實驗提取溶劑進行了研究。
1.2.3.2 前處理方式的選擇 為了優化較好的前處理凈化手段,分別比較直接提取、中性氧化鋁柱[18]和氨基柱三種前處理方式進行比較。并對三種方式最終提取液的總離子流圖及基質效應進行研究來比較凈化效果。
1.2.3.3 樣品除脂探索 參考文獻[8]方法,以辣椒油和火鍋底料為對象,考察經過正己烷脫脂和不脫脂對目標物回收率的影響。
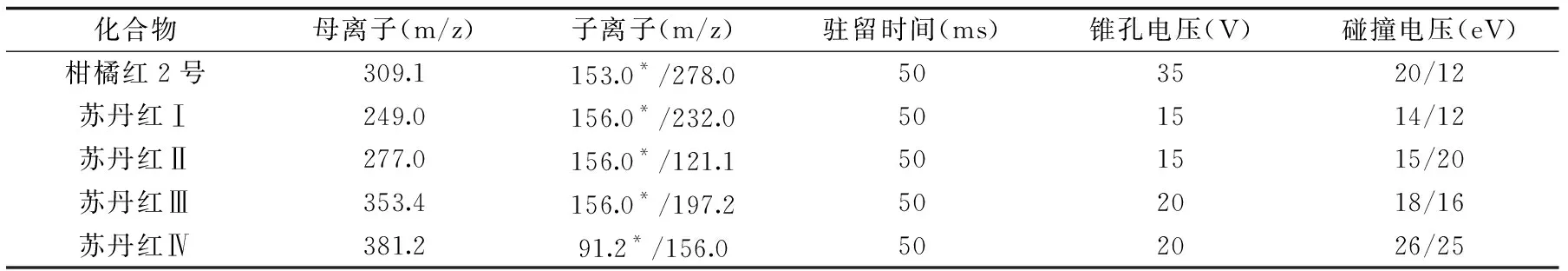
表1 5種非法染料的質譜參數Table 1 Optimized parameters of MS/MS for five illegal dyes
注:*為定量離子。 1.2.3.4 定容體積的選擇 以臍橙、蜜餞、辣椒油、火鍋底料及香辣肉灌腸等基質為研究對象,每類基質添加50 μg/kg含量水平的標準物質,按照1.2.3步驟進行前處理,每個基質做3個平行,定容體積分別為5、10和15 mL,考察不同定容體積對目標物響應及基質效應的影響。
1.2.4 樣品前處理
1.2.4.1 樣品制備 固體樣品(臍橙、蜜餞、香辣肉灌腸)切小塊,以10000 r/min轉速粉碎5 min,充分粉碎、混勻,其中水果樣品全果粉碎;半固體樣品(火鍋底料)直接用粉碎機以5000 r/min轉速,粉碎5 min,粉好的樣品分裝于潔凈容器中;液體樣品(辣椒油)搖勻后備用。
1.2.4.2 樣品提取及凈化 稱取1.2.4.1中制備的樣品2 g(精確至0.01 g)于50 mL離心管中,加入2 g氯化鈉,渦旋1 min,加入10 mL乙腈,渦旋1 min,超聲20 min,以8000 r/min轉速離心3 min。取上清液乙腈層移入15 mL離心管中,40 ℃氮吹干,加入3 mL乙腈-二氯甲烷(1+99)溶液復溶,待凈化。
用6 mL乙腈+二氯甲烷(1+99,體積比)活化氨基柱,待液面降至柱床時,將待凈化液轉移至固相萃取柱,同時收集流出液。用3 mL乙腈+二氯甲烷(1+99,體積比)清洗離心管兩次,淋洗并收集流出液。合并流出液,40 ℃氮氣流下濃縮至干,以甲酸-乙腈(1%)溶液復溶,并定容至10 mL,過微孔濾膜后,供液相色譜-串聯質譜儀測定。
1.2.5 標準溶液的制備 稱取5份相同的空白基質樣品,分別加入一定濃度的標準儲備溶液(1.2.1.1),按1.2.4.2的步驟制備得到含量為2、4、10、20、40 ng/mL的基質配標,得到本實驗所用的基質標準溶液。
1.2.6 線性范圍、決定系數及定量限 稱取5份相同的空白基質樣品,分別加入一定濃度的標準儲備溶液(1.2.1.1),按1.2.4.2的步驟制備得到含量為2、4、10、20、40 ng/mL的基質標準系列工作溶液。將標準工作溶液按濃度由低到高依次測定,以各物質定量離子對的峰面積(Y)對其質量濃度(X)作標準曲線。分別在空白基質中加10、5 μg/kg的目標物,按1.2.4前處理方法處理后,測試其信噪比,分別確定定量限和檢出限。
1.2.7 回收率和精密度 分別在臍橙、蜜餞、辣椒油、火鍋底料和香辣肉灌腸5種基質的陰性樣品中進行10、50、100 μg/kg三個濃度水平的柑橘紅2號和蘇丹紅的加標回收和精密度實驗,每個濃度水平重復測定6次。
1.2.8 基質效應實驗 取空白基質樣品,按1.2.4的步驟制備空白基質溶液。分別用空白溶液和定容溶劑稀釋標準儲備液,得到同濃度水平的測定液。通過分別測定樣品空白提取液與純溶劑中添加同水平目標成分的響應值,計算二者的相對比值來評價基質效應(Matrix effect,ME),ME(%)=(基質匹配標準溶液響應/無基質標準溶液響應-1)×100。基質效應為負值表示存在基質抑制效應;基質效應為正值表示存在基質增強效應,0為無基質效應,絕對值越大,基質效應越強。按所述方法對5種基質中5種非法染料的基質效應進行評價。
1.2.9 實際樣品的測定 在本文建立的試驗條件下,對臍橙、蜜餞、火鍋底料、辣椒油、香辣肉灌腸各10批次共50批次市售實際樣品進行測定。
2 結果與分析
2.1 儀器條件的優化
2.1.1 質譜解析及條件的優化 從圖1的二級全掃描質譜圖可以看到,在正離子模式下,柑橘紅2號及蘇丹紅染料主要以[M+H]+離子形式存在。分析5種非法染料的碎片離子指紋圖譜發現,這幾種偶氮染料具有共同的幾種碎裂方式,均易在偶氮處發生斷裂而得到不同的碎片。首先,該類化合物母體骨架上的酚羥基容易斷裂得到相應的碎片離子[M-OH+H]+;與萘母體上的偶氮在N=N處斷產生具有母核碎片離子156[M-R]+和其取代基碎片離子,其中m/z 156碎片部分為偶氮染料的母體骨架部分,可以作為偶氮類染料的共同特征碎片;此外,Sudan Ⅲ和 Sudan Ⅳ在取代基上的偶氮結構仍可以發生斷裂而得到不同的碎片;柑橘紅2號在其取代基上的醚鍵也容易發生斷裂掉甲氧基碎片,5種偶氮非法染料的二級全掃描質譜圖見圖1。為了同時滿足定性定量的目的,MRM掃描模式下得到5種非法染料的一個母離子和兩個子離子。
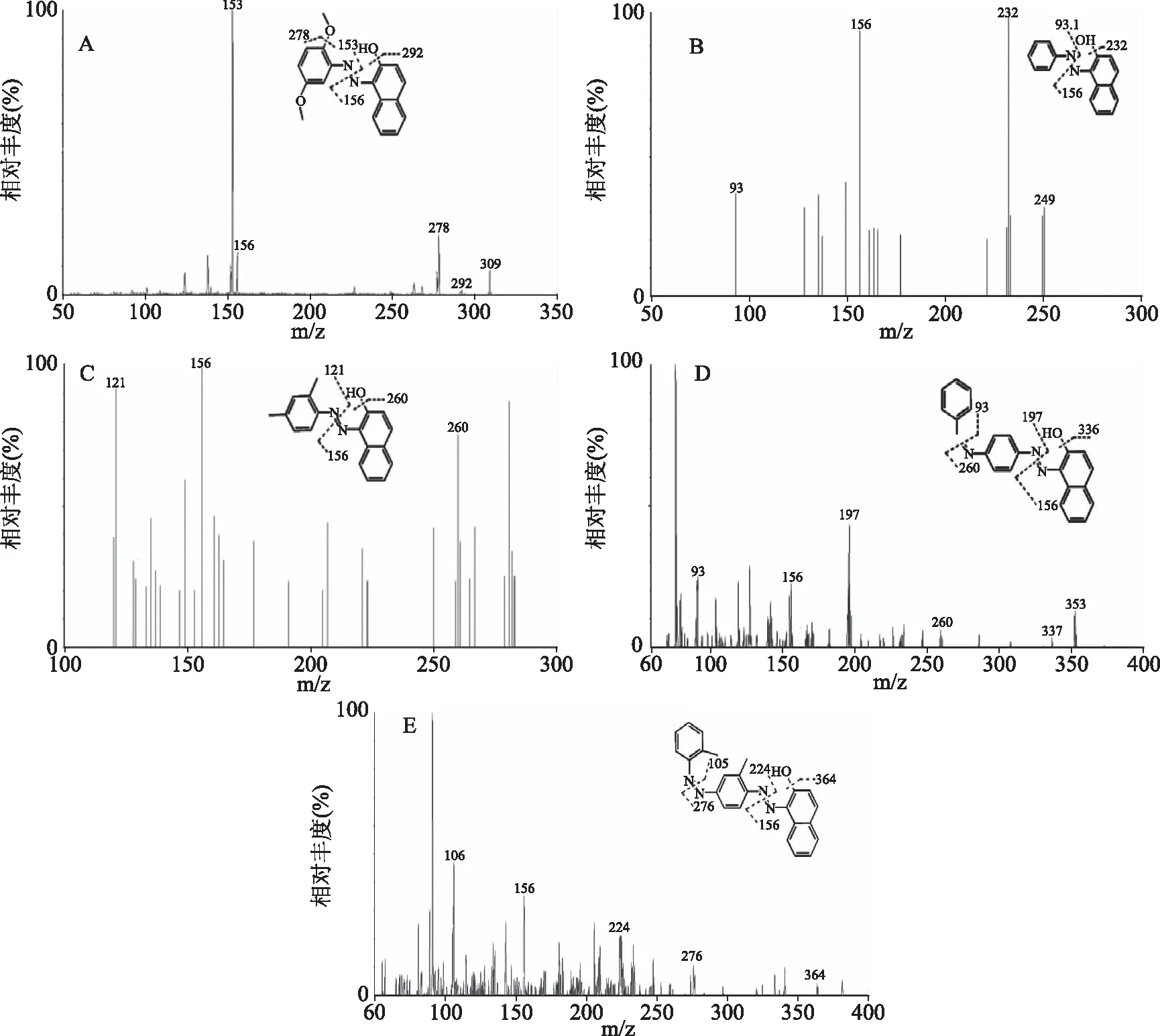
圖1 柑橘紅2號和蘇丹紅Ⅰ~Ⅳ的二級全掃描質譜圖Fig.1 MS2 spectra of Citrus Red 2 and Sudan I~Ⅳ dyes注:A:柑橘紅2號;B:蘇丹紅I;C:蘇丹紅Ⅱ;D:蘇丹紅Ⅲ;E:蘇丹紅Ⅳ。
2.1.2 色譜條件的優化 選用BEH C18色譜柱,分別以考察甲醇-水、乙腈-水、乙腈-10 mmol/乙酸銨溶液和乙腈-0.1%甲酸水溶液幾種流動相條件下目標物的響應,結果發現,乙腈較甲醇作為流動相時,5種染料響應值增強,且峰形更好。而加入0.1%甲酸水溶液可以提高5種染料的離子化效率,并較好地改善峰形。最終確定采用乙腈-0.1%甲酸水作為流動相。通過優化梯度洗脫程序,使目標物與雜質組分有效地分離,且峰形良好。
2.2 前處理條件的優化
2.2.1 樣品提取劑的選擇 考慮到5種偶氮染料的親脂性特點和各種食品等基質較復雜等原因,采用乙腈提取,氯化鈉鹽析,一方面可以有效提高高油脂樣品中偶氮染料的提取效率,另一方面對于含水較多的柑橘樣品,可以促進目標物進入乙腈層,起到鹽析的作用。本方法采用乙腈-氯化鈉提取體系,氨基柱凈化,較好地除去復雜基質中各種高油脂、高糖、高鹽等雜質干擾,結合液相色譜-三重四級桿串聯質譜,可達到檢測要求。
2.2.2 前處理方式的選擇 分別比較直接提取、中性氧化鋁柱[18]和氨基柱三種前處理手段。在前處理過程中發現,對于高油脂等樣品,直接提取和中性氧化鋁柱不能很好地去除油脂,最后存在明顯的油脂殘留。此外,通過三種前處理方式的總離子流圖(見圖2)發現,直接提取方法干擾成分最多,中性氧化鋁柱雖然起到了一定的凈化效果,但是相比氨基柱還是要差一些。另一方面,對三種不同的前處理方式的基質效應進行評價(評價方法見1.2.8),發現中性氧化鋁柱法基質效應與直接提取法相當(基質效應在60%以上),原因可能是,中性氧化鋁法是采用非極性有機溶劑進行提取,對于高油脂樣品中的油脂成分提取率過高,帶來較嚴重的基質效應。通過乙腈提取后經過氨基柱凈化,回收率滿足要求,干擾物較少,基質效應小。氨基柱是以硅膠為基質的氨丙基萃取柱,在非極性有機溶劑中具有強的正相保留作用和弱的陰離子交換雙重保留作用,特別適合于結構相近的化合物的凈化。最終,選擇氨基柱進行凈化。
實驗分別考察了在上樣、淋洗等步驟目標物的保留情況,發現在上樣階段目標物即有10%左右的損失。因此本實驗從上樣開始收集,并用乙腈+二氯甲烷(1+99,體積比)繼續洗脫兩個柱體積(6 mL),結果目標物回收率滿足要求,即洗脫完全。
2.2.3 樣品除脂探索 對于高油脂樣品辣椒油和火鍋底料進行乙腈飽和正己烷脫脂步驟,結果發現,經正己烷脫脂步驟后,目標物回收率降低10%~20%。原因可能是,作為脂溶性較強的偶氮類染料易溶于正己烷,而導致一定的損失。因此,本實驗不進行脫脂步驟。
2.2.4 定容體積 實驗發現,隨著定容體積的增大,基質效應逐漸減小,目標物響應值在定容體積由5 mL變為10 mL時基本沒有變化,當定容體積增大到15 mL時開始有所降低。這是因為,增大定容體積,樣品基質中雜質組分隨之變小,使得基質效應明顯降低,儀器響應提高。另一方面,隨著定容體積增大,樣品中目標物測試濃度降低。
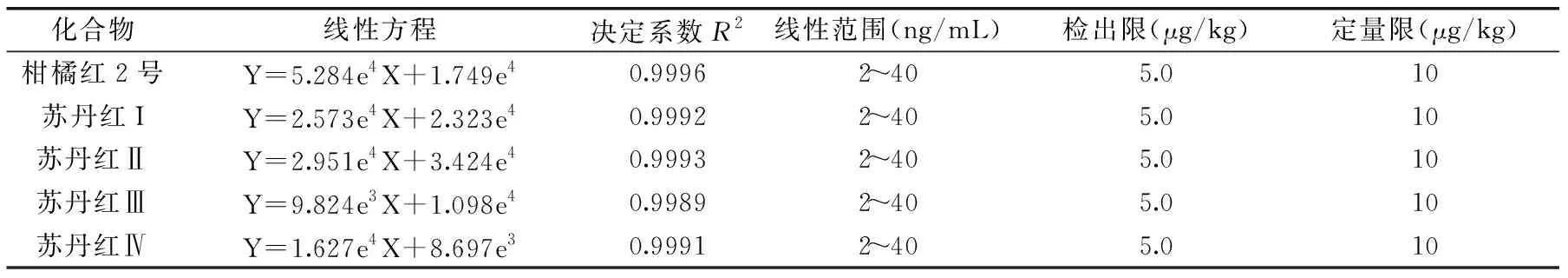
表2 5種非法染料回歸方程、決定系數、線性范圍、檢出限及定量限Table 2 Linear equation,determination coefficient,linear range,limits of detection and quantitation of five illegal dyes
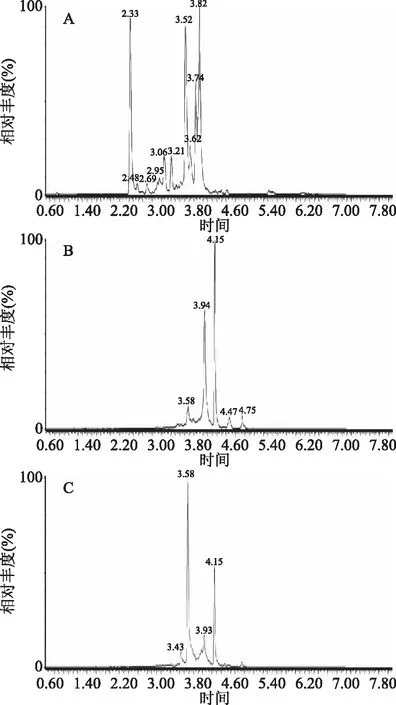
圖2 三種前處理方式下柑橘紅2號和蘇丹紅I~Ⅳ的總離子流圖Fig.2 TIC chromatograms of Citrus Red 2 and Sudan I~Ⅳdyes by three pretreatment methods注:A:直接提取;B:氨基柱凈化;C:中性氧化鋁凈化。
因此本方法確定在2~40 ng/mL線性范圍內定容體積為10 mL。但是,對于超出線性范圍的陽性樣品,可以根據需要增加稀釋倍數。
2.3 線性范圍、決定系數及定量限
將1.2.4步驟制備的標準工作溶液按濃度由低到高依次測定,以各物質定量離子對的峰面積(Y)對其質量濃度(X)作標準曲線,其線性決定系數均大于0.998。分別在空白基質中加10和5 μg/kg的目標物,按1.2.2前處理方法處理后,測試其信噪比,分別確定定量限和檢出限。以信噪比S/N≥10得到目標物的定量限(LOQ),以信噪比S/N≥3得到目標物的檢出限(LOD),結果見表2。
2.4 回收率和精密度
結果顯示在陰性樣品中添加10、50、100 μg/kg三個濃度水平的目標物,其加標回收率在80.7%~114%范圍內,重復測定6次相對標準偏差均小于8.6%,如表3所示。所附的色譜圖是選擇基質復雜的辣椒油為代表,其空白樣品及加標樣品(10 μg/kg)的質量色譜圖見圖3。
2.5 基質效應評價
超高效液相色譜-串聯質譜具有較高的靈敏度,但基于其檢測原理,存在一定的基質效應,特別是對于油脂和蛋白含量高的等辣椒油和肉制品等樣品會存在嚴重的基質效應。建立HPLC-MS/MS方法的同時對該方法的基質效應進行評價,同時采取一定的消除或減弱措施是十分必要的。
結果發現,在5類性質差異較大的基質中,柑橘紅2號及蘇丹紅Ⅰ~Ⅱ的ME值基本在20%以下,基質效應影響較小;蘇丹紅Ⅲ和Ⅳ在蜜餞和辣椒油中存在一定的基質效應,見圖6。鑒于對基質效應的考察,本實驗采用基質匹配校正曲線定量(見1.2.5),以減少基質干擾對結果的影響。
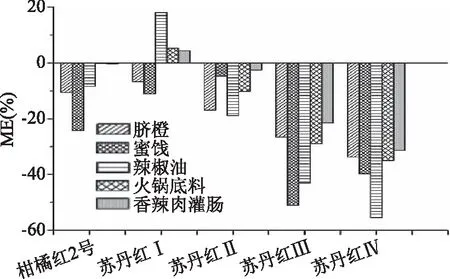
圖4 柑橘紅2號和蘇丹紅Ⅰ~Ⅳ在5種食品中HPLC-MS基質效應評價圖Fig.4 Matrix effects in HPLC-MS analysis of Citrus Red 2 and Sudan I~Ⅳ dyes in five matrics
2.6 實際樣品測定
對50批次市售實際樣品進行測定,包括臍橙、蜜餞(山楂卷)、辣椒油、火鍋底料(紅油半固體)及香辣肉灌腸等樣品,均未檢出含有5種非法染料。
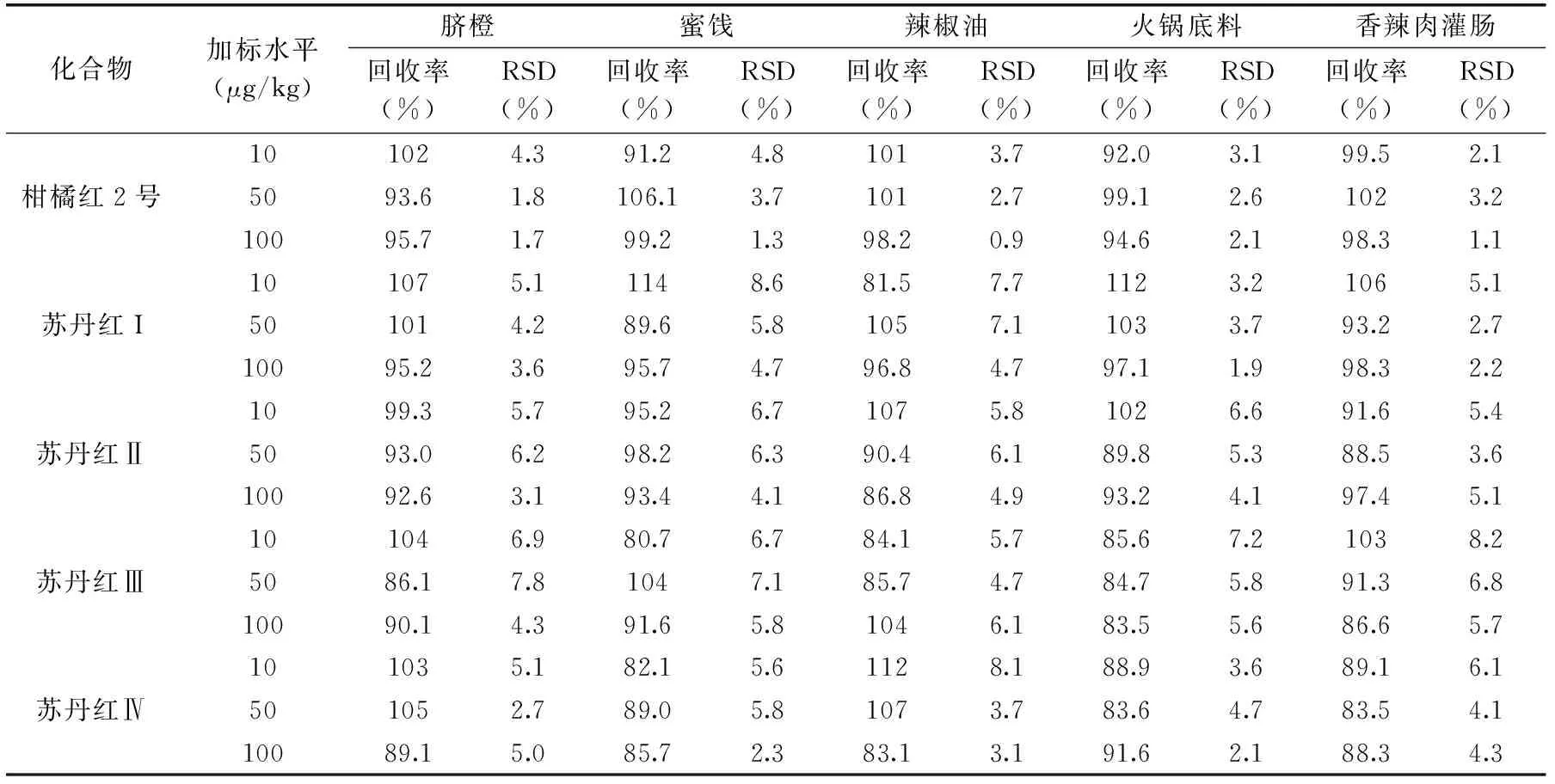
表3 5種基質樣品中5種非法染料添加回收率及精密度(n=6)Table 3 Average recoveries and RSDs of five illegal dyes in five sample matrices(n=6)
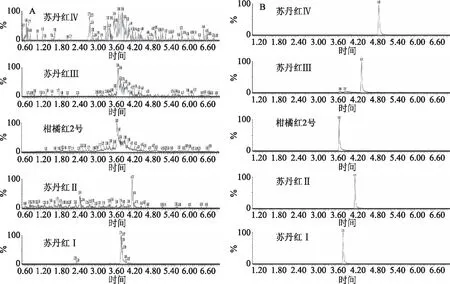
圖3 辣椒油空白樣品及加標樣品(10 μg/kg)中五種非法染料的MRM色譜圖Fig.3 MRM chromatograms of five illegal dyes in chili oil注:A:空白樣品;B:加標樣品(10 μg/kg)。
3 結論
本研究建立了超高效液相色譜-串聯質譜法同時測定食品中柑橘紅2號和蘇丹紅染料的方法。采用乙腈提取,氨基固相萃取富集凈化,高效液相色譜-三重四級桿串聯質譜儀檢測,建立了同時檢測水果、果醬、辣椒制品、火鍋底料及肉制品中柑橘紅2號和蘇丹紅Ⅰ~Ⅳ 5種染料的方法。柑橘紅2號和蘇丹紅Ⅰ~Ⅳ 5種非法染料在2~40 ng/mL范圍內線性關系良好,決定系數(R2)為0.998,回收率在80.7%~114%之間,相對標準偏差RSD<8.6%。方法檢出限為5.0 μg/kg,定量限為10.0 μg/kg,結果準確可靠,簡單易行、靈敏度高、選擇性好,能夠滿足多種食品中非法染料的食品監管需求,適用于食品中痕量水平的非法染料的定性、定量檢測。
猜你喜歡
核科學與工程(2021年4期)2022-01-12 06:30:26
今日農業(2020年19期)2020-12-14 14:16:52
小學生必讀(中年級版)(2020年9期)2020-12-04 02:07:22
科學大眾(2020年17期)2020-10-27 02:49:10
紅土地(2018年11期)2018-12-19 05:10:56
意林·全彩Color(2018年9期)2018-11-13 22:49:38
中學物理·高中(2016年12期)2017-04-22 11:53:03
中國衛生(2016年4期)2016-11-12 13:24:14
中國衛生(2014年4期)2014-12-06 05:57:14
小櫻桃·童年閱讀(2014年11期)2014-12-01 22:21:30
