咖啡酸苯乙酯固體分散體的制備
2019-01-25 07:30:32張德輝龐贊明
中成藥 2019年1期
王 蘭, 張德輝, 沈 文, 龔 頻, 龐贊明, 孫 蕊, 敖 芬
(陜西科技大學, 陜西 西安710021)
咖啡酸苯乙酯作為蜂膠提取物的主要成分之一[1-2], 在抗腫瘤、 保肝、 抗缺血再灌注損傷、 抑菌、 降血糖等方面有不同程度的作用[3-8], 而且迄今尚未發現它對正常細胞的有害影響[1], 故在食品、 醫學等領域具有廣闊的應用前景。 但咖啡酸苯乙酯溶解度很低, 從而影響其生物利用度[1,9], 限制了在治療和預防上的應用, 同時對該成分的研究目前主要集中在提取工藝、 藥理活性等方面, 鮮有制劑方面的報道。
研究表明, 固體分散體可提高水溶性差的藥物的溶 解 度[10]。 因 此, 本實 驗 以PVP K30 為 載體[11], 采用溶劑法制備咖啡酸苯乙酯固體分散體,并對其溶解度及體外溶出特性進行研究, 然后通過掃描電鏡、 X-射線粉末衍射法、 差示掃描量熱法對其進行物相鑒別。
1 材料
1.1 試藥 咖啡酸苯乙酯原料藥(湖北遠成賽創科技有限公司); 咖啡酸苯乙酯對照品(中國食品藥品檢定研究院); 聚乙烯吡咯烷酮K30 (PVP K30, 上海藍季生物有限公司)。
1.2 儀器 TA2603B 電子天平(上海天美天平儀器有限公司); ZRS-8GD 智能溶出試驗儀(天津市天大天發科技有限公司); UV-5100 紫外可見分光光度計(上海元析儀器有限公司); RE52CS 旋轉蒸發儀(上海亞榮生化儀器廠); X-射線粉末衍射儀; 飛納Phenom Pro 臺式掃描電鏡; STA409PC 同步綜合熱分析儀(德國耐馳公司)。
2 方法與結果
2.1 檢測波長選擇 無水乙醇配制質量濃度適宜的咖啡酸苯乙酯溶液, 再配制空白輔料, 過0.45 μm微孔濾膜, 在200 ~400 nm 波長范圍內進行紫外掃描。結果, 咖啡酸苯乙酯在330 nm 波長處有最大吸收, 空白輔料無干擾, 故選擇其作為檢測波長。
2.2 線性關系考察 精密稱取咖啡酸苯乙酯對照品1 mg, 無水乙醇定容于50 mL 量瓶中, 得到20 μg/mL貯備液, 分別精密量取2、 3、 4、 5、 6、7、 8、 9 mL 于10 mL 量瓶中, 定容至刻度。 以吸光度為縱坐標(Y), 咖啡酸苯乙酯質量濃度為橫坐標(X) 進行回歸, 得到回歸方程為Y =0.056 9X +0.071 8 (R2=0.999 7), 在4~18 μg/mL 范圍內線性關系良好。
2.3 精密度、 準確度試驗 配制低、 中、 高(4、10、 15 μg/mL) 質量濃度的咖啡酸苯乙酯溶液,同1 d 內平行測定5 次, 考察日內精密度; 每天測定1 次, 連續5 d, 考察日間精密度。結果, 低、中、 高質量濃度溶液的日內、 日間精密度RSD 分別為1.12%、 1.42%, 1.23%、 1.89%, 0.97%、1.51%, 準確度分別為97%、 103%、 96%, 表明該方法精密度、 準確度良好。
2.4 樣品制備
2.4.1 固體分散體 按處方比例(1 ∶4、 1 ∶6、1 ∶8、 1 ∶10、 1 ∶12) 分別稱取適量咖啡酸苯乙酯、 PVP K30, 溶于適量無水乙醇中, 混合均勻后置于旋轉蒸發儀上, 60 ℃下水浴旋蒸至黏稠狀態,轉移至烘箱中烘干, 粉碎, 過80 目篩, 即得, 置于干燥器中保存備用。
2.4.2 物理混合物 按“2.4.1” 項下處方比例分別稱取適量咖啡酸苯乙酯、 PVP K30, 研細后等量遞加法混合均勻, 過80 目篩, 即得, 置于干燥器中保存備用。
2.5 溶解度測定 取適量咖啡酸苯乙酯、 固體分散體, 置于100 mL 燒杯中, 加50 mL 水, 在(25±1)℃下磁力攪拌4 h (吸光度不再變化時達到平衡), 使其成為過飽和溶液, 0.45 μm 微孔濾膜過濾, 取續濾液, 計算兩者在水中的平衡溶解度。
2.6 溶出度測定 按照2015 年版《中國藥典》四部通則0931 “溶出度與釋放度測定法” 第二法(槳法) 測定[12], 以900 mL 脫氣水為溶出介質,滿足漏槽條件。 投入15 mg 咖啡酸苯乙酯及各待測處方載藥量均為15 mg 的固體分散體, 在溫度(37.0±0.5)℃、 轉速100 r/min 條件下于10、 20、30、 40、 50、 60 min 取樣5 mL, 同時補充等溫同體積溶出介質, 樣品過0.45 μm 微孔濾膜后測定吸光度, 并代入“2.2” 項下回歸方程, 測得各時間點質量濃度, 計算累積溶出度, 繪制溶出曲線。
2.7 載體材料選擇 在預實驗中選擇PEG6000、PVP K30 作為載體材料制備固體分散體, 發現采用熔融法時, PEG6000 制備的固體分散體材質偏軟, 不易干燥粉碎, 難以取用; 采用溶劑法時,PVP K30 制備的固體分散體易干燥粉碎, 便于取用。 因此, 選擇PVP K30 作為載體進行下一步實驗, 并通過溶劑法制備固體分散體。
2.8 載體比例選擇
2.8.1 溶出度 圖1 顯示, 相同比例下固體分散體的溶出速率明顯大于原料藥和物理混合物, 而且在10 min 時的累積溶出度最高可達到80% 左右,而物理混合物僅為6%左右, 原料藥更是只有1%,表明PVP K30 對咖啡酸苯乙酯溶出具有一定促進作用。 同時, 咖啡酸苯乙酯與PVP K30 的最佳比例為1 ∶10。
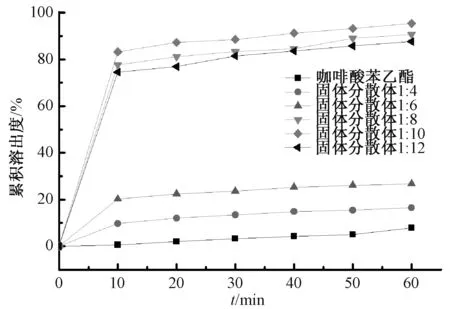
圖1 固體分散體溶出曲線Fig.1 Dissolution curves for solid dispersions
2.8.2 溶解度 圖2 顯示, 25 ℃時咖啡酸苯乙酯的溶解度為(23.374±1.572) μg/mL, 物理混合物略有增加, 而固體分散體顯著提高, 以咖啡酸苯乙酯與PVP K30 比例1 ∶10 時最佳, 比原料藥提高了56%, 但與其他比例相比提升不明顯, 而且隨著其增加載藥量反而降低。 因此, 選擇兩者比例為1 ∶10。
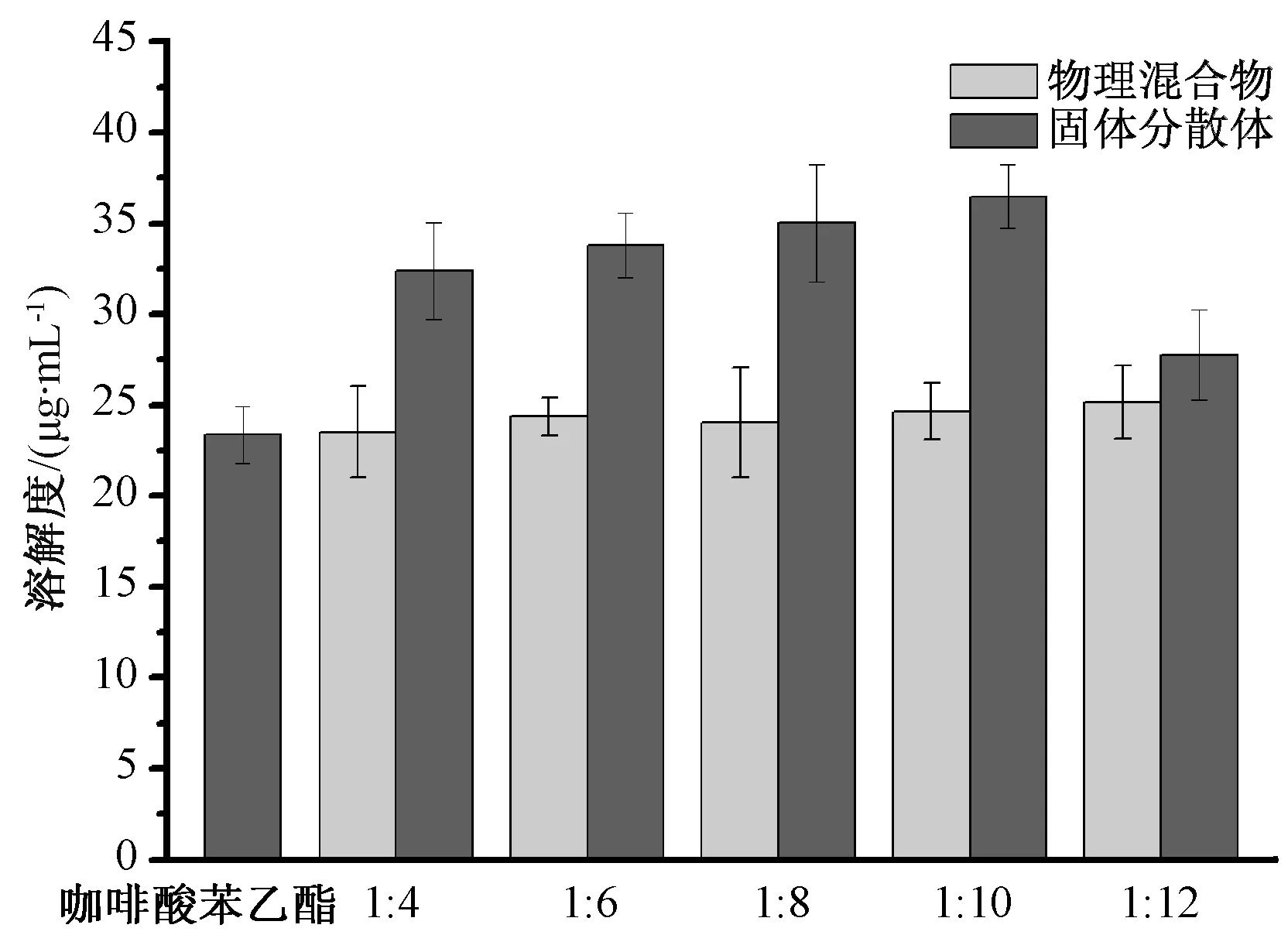
圖2 固體分散體溶解度Fig.2 Solubilities of solid dispersions
2.9 掃描電鏡(SEM) 條件為真空鍍金60 s,高壓5 kV, 采用SEM 觀察樣品形態和晶體結構,結果見圖3。 由圖可知, 咖啡酸苯乙酯以大小不一的矩形片狀晶體形式存在; PVP K30 為球形或類球形, 物理混合物中仍可觀察到咖啡酸苯乙酯結構和PVP K30 存在, 而且兩者互無影響; 固體分散體中已無明顯咖啡酸苯乙酯晶體結構存在, 表明該成分以非結晶態均勻分散在PVP K30 中, 并可能已形成固體分散體。
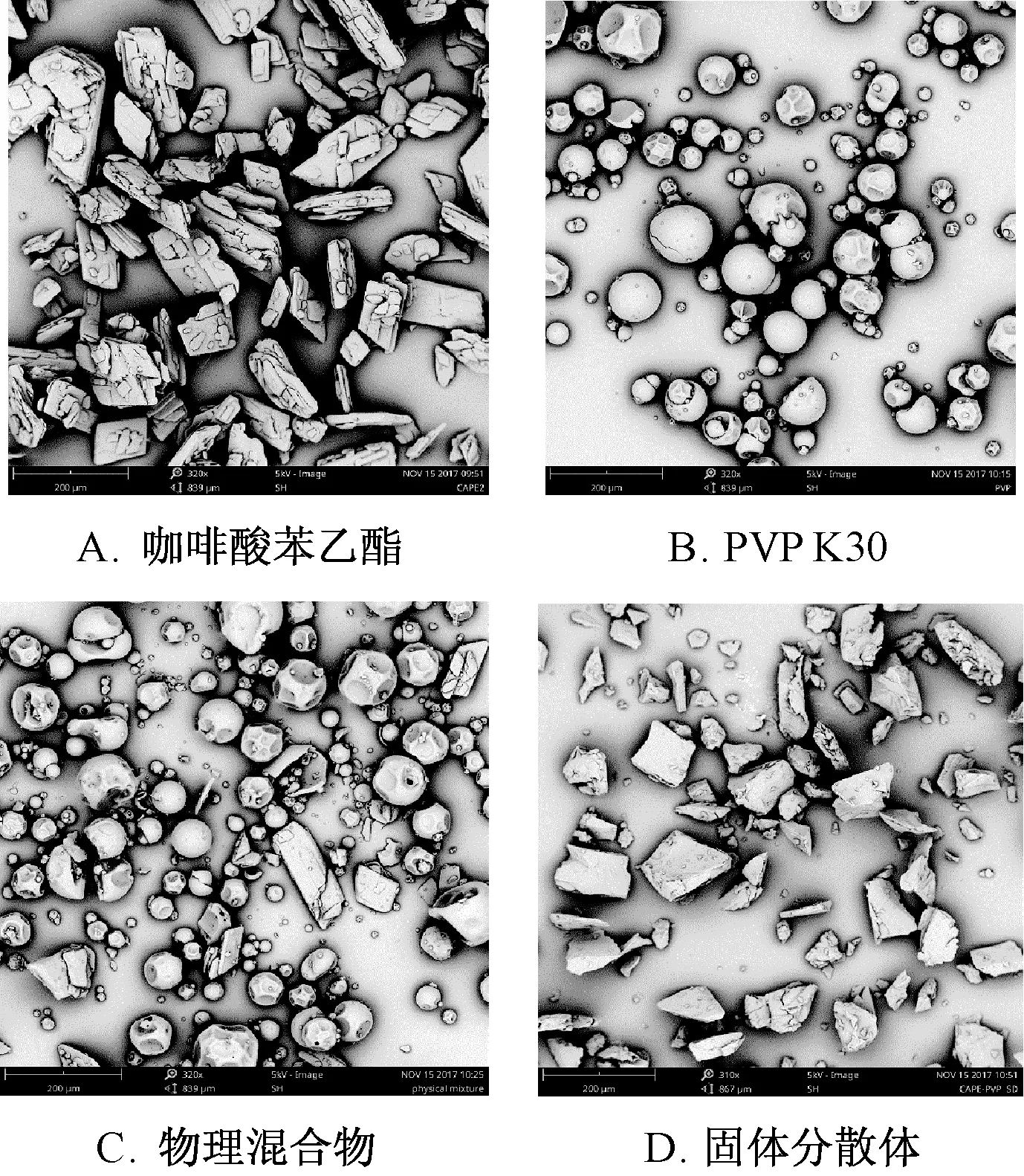
圖3 樣品SEM 圖Fig.3 SEM images for samples
2.10 差示掃描量熱(DSC) 以空白鋁坩堝為參比物, N2為保護氣, 溫度40 ~150 ℃, 升溫速度5 ℃/min, 采用DSC 對樣品進行分析, 結果見圖4。 由圖可知, 咖啡酸苯乙酯在128 ℃處有1 個熔融吸熱峰, 為其熔點, 與前期報道[1]一致; PVP K30 在128 ℃處未出現明顯吸收峰; 物理混合物在128 ℃處仍存在較小的熔融吸熱峰, 為咖啡酸苯乙酯熔點, 表明PVP K30 與其未發生相互作用, 該成分仍以晶體形式存在; 固體分散體在128 ℃處吸收峰消失, 表明咖啡酸苯乙酯與PVP K30 可能形成低共熔物或共沉淀物, 即以無定形態分散在載體中。
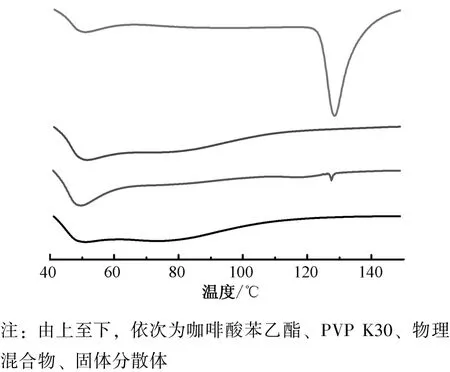
圖4 樣品DSC 曲線Fig.4 DSC curves for samples
2.11 X 射線衍射(XRD) 采用XRD 對樣品進行分析, 結果見圖5。 由圖可知, 咖啡酸苯乙酯在6.24°處有較強的吸收峰, 表明它以結晶態存在;PVP K30 在0°~10°處無特征衍射峰, 表明它是無定型材料; 物理混合物在6.24°處的峰強度減弱,表明咖啡酸苯乙酯仍以晶體形式存在, 但占總成分比例降低, 故其峰強度低于咖啡酸苯乙酯; 固體分散體衍射峰消失, 表明咖啡酸苯乙酯以無定形狀態存在于PVP K30 中。
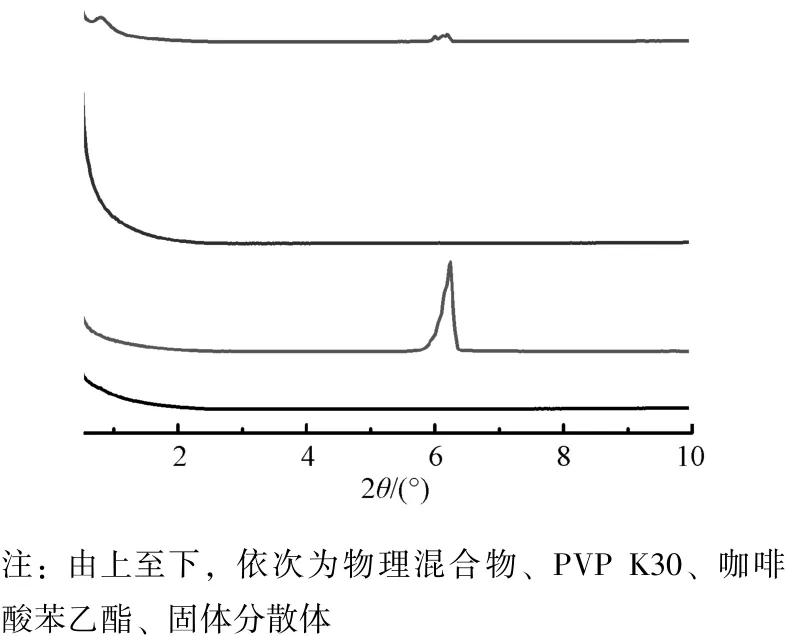
圖5 樣品XRD 圖Fig.5 XRD patterns for samples
3 討論
本實驗制備了咖啡酸苯乙酯固體分散體, 發現其溶解度及溶出速率與原料藥相比得到了顯著提高。 結合SEM、 XRD、 DSC 分析可知, 載體與藥物共蒸發溶劑時, 藥物分子以無定形狀態分散于載體中, 兩者之間存在氫鍵等相互作用, 導致在固化時形成了具有較高能量的非結晶無定形物, 從而增加咖啡酸苯乙酯在溶出介質中的釋放速率。 另外,咖啡酸苯乙酯與PVP K30 比例為1 ∶10 時, 所得固體分散體中前者溶解度、 溶出度最大。
綜上所述, 本實驗提供了一種簡單有效的咖啡酸苯乙酯固體分散體制備方法, 它作為一種藥物中間體, 可為片劑、 顆粒劑等劑型的研制奠定基礎。
