達烏里胡枝子沒食子酸提取條件的優化
2019-03-15 09:00:00趙宇星李小鵬
山西農業科學 2019年3期
趙宇星,李小鵬,任 敏,趙 祥
(山西農業大學動物科技學院,山西太谷030801)
植物酚類物質是由植物的次生代謝途徑產生的,其在植物體內分布廣泛、種類繁多,但是對植物的初級生產沒有直接影響,而是作為一種次生防御物質發揮著重要的作用[1]。酚類物質是植物體內普遍存在的次生代謝物,廣泛存在于植物的葉、根、皮、花和果實中[2]。酚酸類物質對多種植物病原真菌均可抑制活性[3]。達烏里胡枝子(Lespedeza davurica)是豆科(Leguminosae)蝶形花亞科(Papilionatae)胡枝子屬(Lespedza Michx.)的喜暖旱中生半灌木,其含有豐富的酚類物質[4],干旱脅迫能夠顯著提高達烏里胡枝子的抗氧化能力[5]。沒食子酸(Gallic acid,GA)又稱五倍子酸,是一種天然酚類化合物[6],能夠抗菌、抗突變、消除自由基[7]。因此,沒食子酸在提高植物適應極端環境方面發揮著重要作用。
目前,胡枝子屬植物不僅被廣泛用于各種動物的飼草,而且在藥用領域也有著廣泛的應用[8];已從達烏里胡枝子中分離鑒定了香葉木素、山奈酚、檉柳素、木犀草素、槲皮素等11種化合物[4]。達烏里胡枝子的研究多集中在自然狀態下胡枝子體內的單寧含量,但是對達烏里胡枝子酚類次生代謝物質的研究較少。
本試驗以達烏里胡枝子為對象,研究了其沒食子酸最佳的提取工藝及其最優測定條件,旨在為達烏里胡枝子沒食子酸的提取和利用提供依據。
1 材料和方法
1.1 試驗材料
供試材料為晉農1號達烏里胡枝子,由山西農業大學牧站草學實驗室種子資源庫提供。沒食子酸標準品(99%),來源于北京索萊寶公司。
1.2 試驗設計
試驗采用盆栽方式,于2016年4月1日在山西農業大學草業科學系溫室中進行,平均溫度為14.5~29.5℃,相對濕度為64%~79%。土壤來源于試驗田用地過篩耕層土(0~20 cm),與細沙2∶1混合,pH值為7.5,并測得田間最大持水量約為24%。
試驗采用上端口直徑為21 cm、高26 cm的塑料桶,每桶裝風干土約7.5 kg,播種量為55粒/盆。待苗齊后,每桶留苗25株。待達烏里胡枝子長到分枝期取樣放置于裝有冰袋的泡沫箱中,迅速帶回實驗室保存于超低溫-80℃冰箱,以備測定。
1.2.1 達烏里胡枝子單因素提取試驗 設定蒸餾水、60%甲醇、60%乙醇為提取溶劑,其他提取條件為固液比1∶40,超聲時間30 min,超聲溫度40℃;提取溶劑濃度為20%,40%,60%,80%共4個梯度,其他提取條件為固液比1∶50,超聲時間30 min,超聲溫度60℃;固液比為1∶30,1∶40和1∶50,其他提取條件提取溶劑為60%甲醇,超聲時間30 min,超聲溫度60℃;測定時間為30,45,60 min,其他提取條件為提取溶劑為60%甲醇,固液比1∶40,超聲溫度60℃;測定溫度分別設定為40,50,60,70℃,其他提取條件提取溶劑為60%的甲醇,固液比1∶40,超聲時間為30 min,測定達烏里胡枝子沒食子酸含量,確定單因素提取條件。
1.2.2 正交試驗確定沒食子酸的最佳提取條件根據單因素試驗確定的因素和水平,選取溶劑濃度(A)、固液比(B)、提取時間(C)、提取溫度(D)4個因素,采用正交表L9(34)進行試驗,以確定沒食子酸的最佳提取條件(表1)。
1.2.3 沒食子酸含量的測定
1.2.3.1 分析樣品的制備 精確稱取達烏里胡枝子樣品0.200 g(±0.005 g),加入有提取溶劑10 mL的具塞試管中,超聲提取,超聲后離心取上清液,過0.45 μm濾膜,供試驗測定。
1.2.3.2 色譜條件的確定 采用Waters 1500高效液相色譜儀-2489紫外檢測器測定,C18(250 mm×4.6 mm,5 μm)色譜柱,分別選取色譜條件:甲醇-0.1%磷酸[9](色譜條件1);乙腈(A)-0.1%磷酸(梯度洗脫0~10 min,A 2%~10%)[10](色譜條件2);乙腈(A)-0.2%冰醋酸(梯度洗脫 0~10 min,A 5%~12%)[11](色譜條件3);乙腈(A)-0.15%三氟乙酸(梯度洗脫0~10 min,A 0%~7%)[12](色譜條件4);乙腈(A)-0.15%三氟乙酸(梯度洗脫0~5 min,A 0%~3%)[13](色譜條件5),測定達烏里胡枝子沒食子酸含量,以確定色譜條件。
1.2.3.3 沒食子酸標準液相色譜 其色譜圖如圖1所示。
1.2.3.4 標準曲線的繪制 準確稱取沒食子酸對照品20.0mg,甲醇溶解并定容到100mL,得200μg/mL的沒食子酸溶液,以此液為母液,進一步稀釋成濃度梯度為 0.5,1,5,10,15,20 μg/mL 的沒食子酸溶液,分別取20 μL進樣于高效液相色譜儀進行測定,并且以峰面積為縱坐標,沒食子酸含量為橫坐標,繪制標準曲線。
1.2.3.5 穩定性試驗 精確稱取達烏里胡枝子樣品8份各0.200 g,按正交試驗結果中最優條件進行提取,制備提取液,每隔15 min測定一份,記錄數據并計算標準偏差RSD值。
1.3.6 加樣回收率試驗 精確稱取達烏里胡枝子樣品8份各0.200 g,按正交試驗結果中最優條件進行提取,制備提取液,向各個樣品浸提液中分別加入一定量的標樣,測定含量,計算加樣回收率。
1.3 數據分析
所有試驗數據均采用Microsoft Excel 2016和SPSS 22.0進行統計分析。采用單因素方差分析(ANOVA)比較不同處理間達烏里胡枝子沒食子酸含量差異顯著性。使用SigmaPlot 13.0軟件作圖。
2 結果與分析
2.1 達烏里胡枝子沒食子酸最佳提取條件篩選
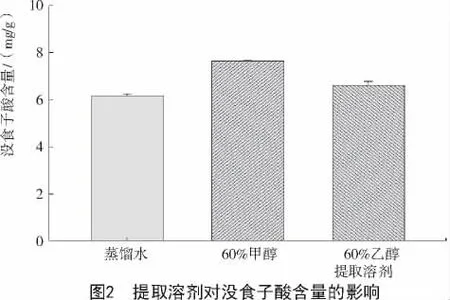
2.1.1 提取溶劑對達烏里胡枝子沒食子酸含量的影響 不同提取溶劑對達烏里胡枝子沒食子酸含量的影響不同(圖2),其中,用60%甲醇提取溶劑提取時,測得沒食子酸含量最高,為7.65 mg/g;用蒸餾水溶劑提取時,測得沒食子酸含量最低,為6.16mg/g。3種溶劑測得沒食子酸含量由高到低順序為60%甲醇>60%乙醇>蒸餾水。因此,選擇60%甲醇為提取的最佳溶劑。
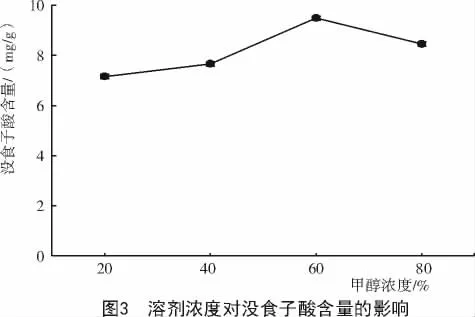
2.1.2 提取溶劑濃度對達烏里胡枝子沒食子酸含量的影響 不同提取溶劑濃度對達烏里胡枝子沒食子酸含量的影響不同(圖3),隨著甲醇濃度的升高沒食子酸含量表現為先升后降的變化趨勢,當甲醇濃度為60%時,沒食子酸含量達最大值,為9.49mg/g;當甲醇濃度超過60%時,沒食子酸含量有明顯降低的趨勢。因此,選擇60%為最佳提取溶劑濃度。
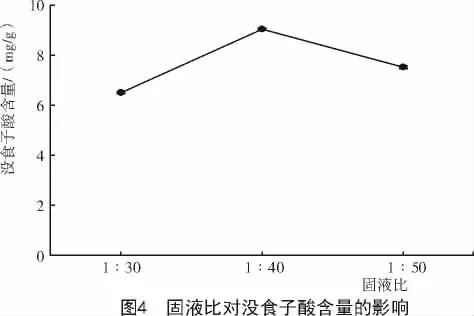
2.1.3 提取固液比對達烏里胡枝子沒食子酸含量的影響 不同固液比對達烏里胡枝子沒食子酸含量的影響不同(圖4),當固液比為1∶40時,沒食子酸含量最高,為9.04 mg/g。因此,選擇1∶40為最佳提取固液比。
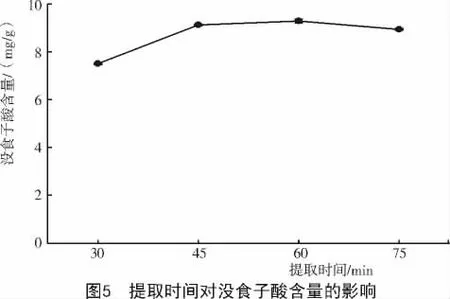
2.1.4 提取時間對達烏里胡枝子沒食子酸含量的影響 不同提取時間對達烏里胡枝子沒食子酸浸出速度的影響不同(圖5),當提取時間為30~45min時,達烏里胡枝子沒食子酸浸出速度較快;當提取時間為45~60 min時,沒食子酸含量浸出速度較慢,且在60 min時沒食子酸含量達到最大值,為9.283 mg/g;當浸提時間超過60 min后,沒食子酸含量開始降低。因此,選擇60 min為最佳提取時間。
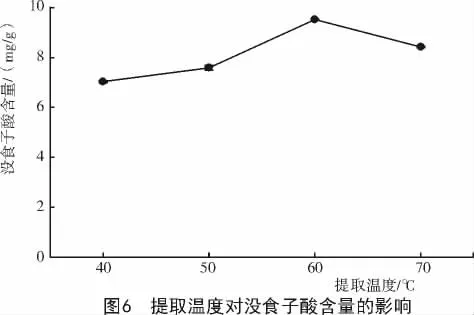
2.1.5 提取溫度對達烏里胡枝子沒食子酸含量的影響 不同提取溫度對達烏里胡枝子沒食子酸浸出速度的影響不同(圖6),隨著溫度的升高,達烏里胡枝子沒食子酸含量增加,當提取溫度超過50℃時,沒食子酸含量增長的幅度較大,且在60℃時達到最大值,為9.523 mg/g;60℃以后溫度繼續升高,沒食子酸含量反而降低,且降低幅度較大。因此,選擇60℃作為最佳提取溫度。
2.2 高效液相色譜測定條件對達烏里胡枝子沒食子酸含量的影響
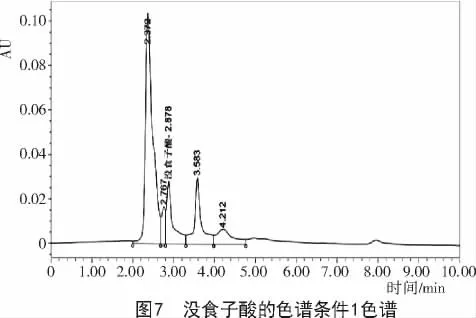
2.2.1 色譜條件1對達烏里胡枝子沒食子酸含量的影響 采用甲醇和0.1%的磷酸作為流動相,甲醇與水的比例為1∶9時,沒食子酸的峰與雜質峰未完全分離(圖7),且洗脫出的其他雜質峰偏多,并未出現較為理想的色譜圖。
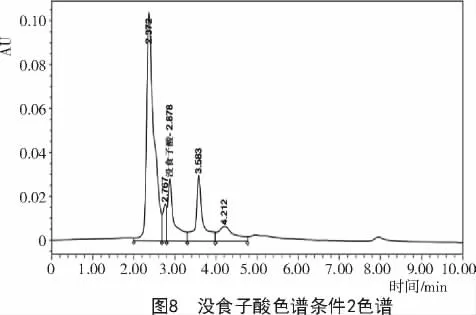
2.2.2 色譜條件2對達烏里胡枝子沒食子酸含量的影響 采用乙腈(A)與0.1%的磷酸(B)作為流動相,梯度洗脫0~10 min,A 2%~10%,體積流量1 mL/min,檢測波長280 nm時,雜質峰仍然較多(圖8),未得到較好的色譜圖。
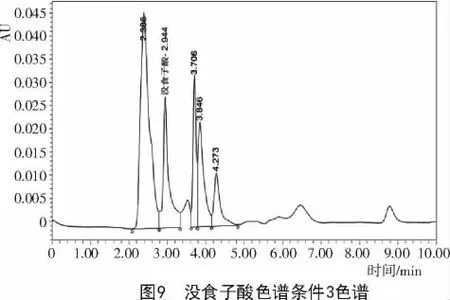
2.2.3 色譜條件3對達烏里胡枝子沒食子酸含量的影響 采用乙腈(A)-2%冰醋酸(B)為流動相,梯度洗脫0~10min,A5%~12%;體積流量1mL/min,檢測波長280 nm,柱溫28℃,雜質峰仍然較多(圖9),并未得到理想的色譜圖。
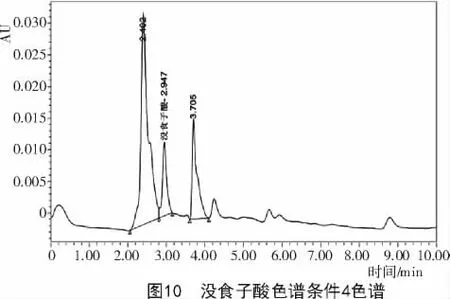
2.2.4 色譜條件4對達烏里胡枝子沒食子酸含量的影響 采用乙腈(A)和0.15%三氟乙酸(B)作為流動相,梯度洗脫0~10min,A0%~7%;10~20min,A7%~16%,體積流量1 mL/min,檢測波長280 nm,柱溫28℃,雜質峰明顯減少,但沒食子酸前后峰保留時間與沒食子酸峰保留時間較近(圖10),未得到較為理想的色譜圖。
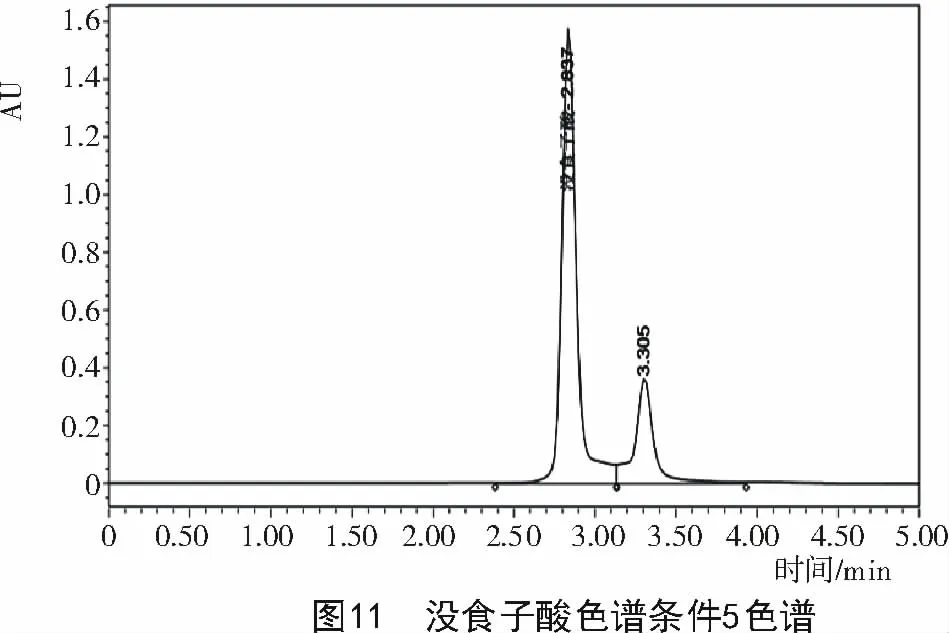
2.2.5 色譜條件5對達烏里胡枝子沒食子酸含量的影響 將色譜條件4優化后,用乙腈(A)和0.15%TFA(B)作為流動相,梯度洗脫變為 0~5 min,A 0%~3%,體積流量1 mL/min,檢測波長280 nm,柱溫30℃,色譜圖如圖11所示,得到了較好的沒食子酸色譜圖。
2.3 綜合提取條件對達烏里胡枝子沒食子酸含量的影響
通過正交試驗可知,綜合影響達烏里胡枝子沒食子酸含量的最佳提取條件4個因素的主次順序為A>B>D>C,即提取溶劑濃度、固液比、提取溫度、提取時間,A2B2C1D2為最佳提取工藝(表2)。結合單因素試驗,根據沒食子酸提取規律,最終選擇溶劑濃度為60%甲醇,固液比為1∶40,提取時間為45 min,提取溫度為60℃。在此條件下,測得達烏里胡枝子沒食子酸的保留時間為2.837 min,沒食子酸色譜峰的理論塔板數均不低于5 000,沒食子酸與相鄰色譜峰的分離度大于1.5,說明沒食子酸與其他雜質色譜峰達到了基線分離(圖11)。
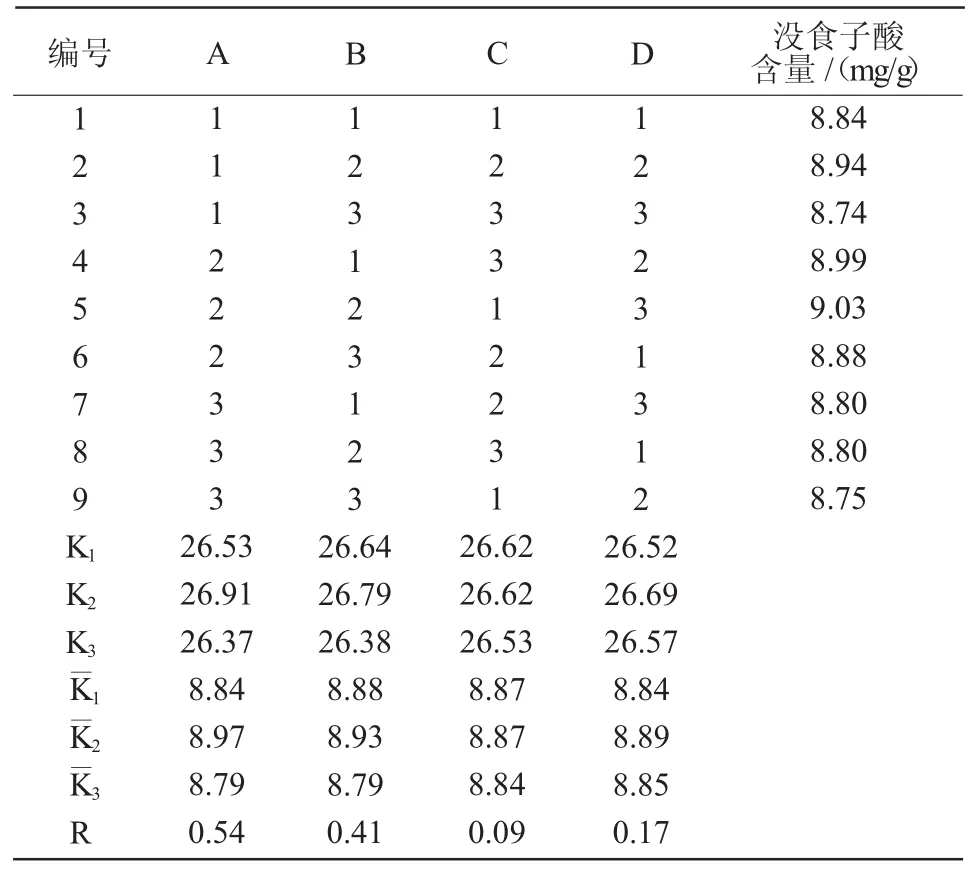
表2 正交試驗確定沒食子酸最佳條件
沒食子酸的峰面積(Y)與沒食子酸的濃度(X)回歸方程為:Y=5.094X+7.594 4(R2=0.999 362),表明沒食子酸在4.91~30.64 μg/mL范圍內,沒食子酸峰面積值與質量濃度之間呈現良好的線性關系。在 0,0.25,0.5,1,2,4,8,24 h 測得達烏里胡枝子沒食子酸含量為7.30~7.73 mg/g,其RSD值為0.021%(表3),表明24 h內供試液穩定性良好。按照已知的方法對樣品液制備處理,且用已知的色譜條件進行沒食子酸含量的測定,最終計算出達烏里胡枝子沒食子酸平均含量為7.57 mg/g,并計算回收率,結果表明,平均回收率為99.58%,RSD為0.30%(表 4)。
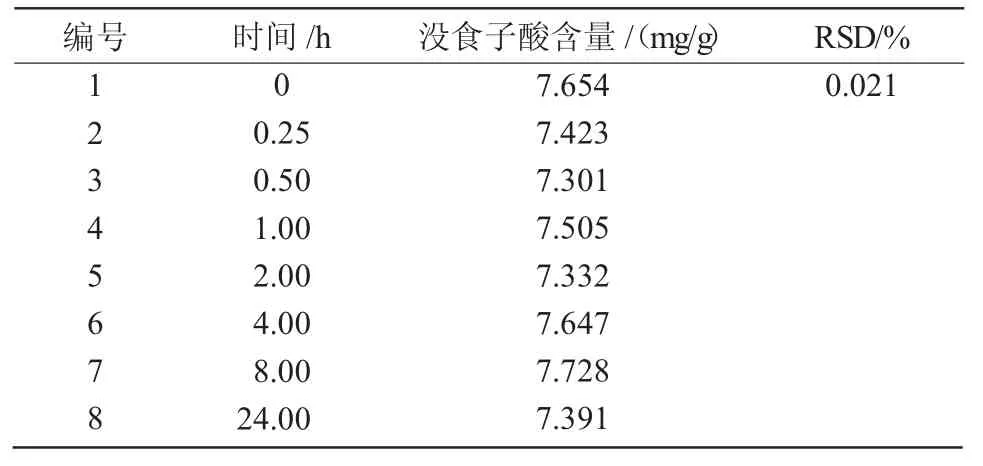
表3 穩定性測定結果
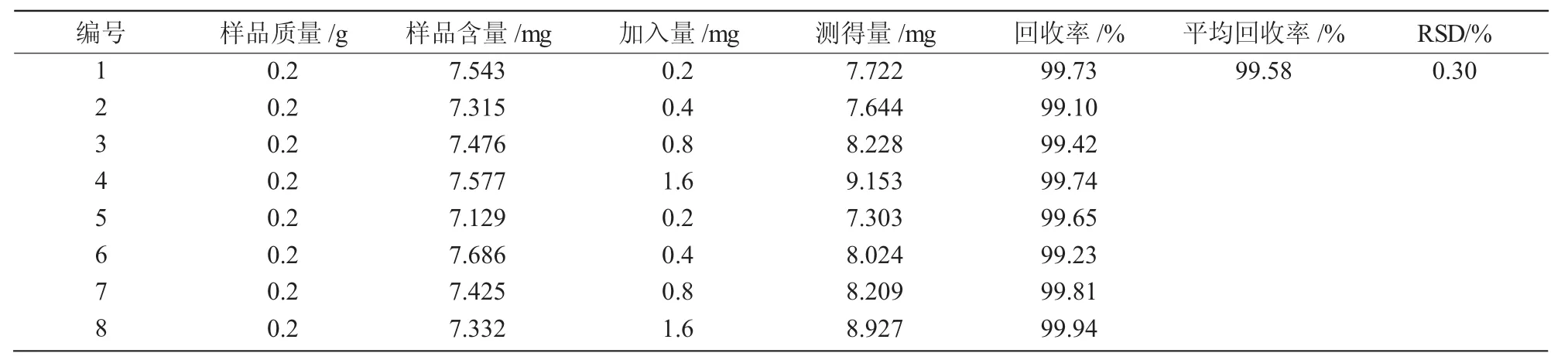
表4 加樣回收率測定結果
3 討論
本研究采用超聲提取法對達烏里胡枝子沒食子酸含量進行測定,以60%甲醇、60%乙醇、蒸餾水作為提取溶劑進行提取,結果發現,60%甲醇提取的沒食子酸含量最高,并選取20%,40%,60%,80%的甲醇分別進行提取,結果表明,60%甲醇提取的沒食子酸含量最高,與關瀟瀅等[13]和金玲等[14]以甲醇作為提取溶劑,沒食子酸含量最高,穩定性最好,峰形對稱,分離度好的結果一致。故選用60%甲醇作為提取溶劑。
本研究通過采用 1∶30,1∶40,1∶50的固液比分別進行提取,結果表明,固液比為1∶40時,提取的沒食子酸含量最高,原因可能是由于在一定的體積范圍內,增加提取液的體積可增大提取液與提取物的接觸面積,使得沒食子酸在甲醇中較多溶出,但隨著提取液體積的增加沒食子酸溶出達到飽和,再增加提取液的體積,沒食子酸的溶出會減小[15]。故選取1∶40作為固液比。
本研究通過采用 30,45,60,75 min 作為提取時間分別進行提取,結果表明,60 min時,沒食子酸含量最高,原因可能是由于60 min之前,沒食子酸溶出不完全;當時間到達60 min時,沒食子酸含量溶出達到最大值;60 min之后,隨著時間的延長,沒食子酸受到氧化的時間延長,沒食子酸含量故而下降[16-17]。因此,選取60 min作為提取時間。
本研究通過采用40,50,60,70℃作為提取溫度分別進行提取,結果表明,60℃時,沒食子酸含量最高,這是由于隨著溫度的升高,分子熱運動速度加快,滲透、擴散、溶解速度加快,使沒食子酸更容易從細胞中轉移到溶劑中,從而使沒食子酸的含量提高;當溫度上升到一定程度時,部分沒食子酸可能被氧化破壞,或者由于沒食子酸分子結構改變,從而使其含量下降[15]。故選取60℃作為提取的溫度。
本研究通過正交試驗,確定達烏里胡枝子沒食子酸的最佳提取工藝,最佳提取條件為溶劑濃度為60%甲醇,固液比為1∶40,提取時間為45 min,提取溫度為60℃,這與黃斌等[17]的研究結果相似。
對沒食子酸對照品甲醇溶液的紫外可見光(190~600 nm)掃描發現,沒食子酸在280 nm處有最大吸收波長。故選擇280 nm為其檢測波長。
流動相分別考察了甲醇-0.1%磷酸、乙腈-0.1%磷酸、乙腈-0.2%冰醋酸、乙腈-0.15%三氟乙酸,結果發現,流動相為乙腈-0.15%三氟乙酸時,峰型和分離度均較好;其他條件下,雜質峰較多,且與沒食子酸峰未完全分離。故選用乙腈-0.15%三氟乙酸作為流動相。
4 結論
本研究通過正交試驗,得到達烏里胡枝子沒食子酸最佳提取條件為溶劑濃度60%甲醇、固液比為1∶40、提取時間為45 min、提取溫度為60℃,在該條件下,采用乙腈-0.15%三氟乙酸梯度洗脫,流量為1 mL/min,檢測波長為280 nm,柱溫30℃。結果表明,沒食子酸在4.91~30.64 μg/mL范圍內呈現良好的線性關系,平均加樣回收率為99.58%,RSD為0.30%;并且利用上述方法,測得達烏里胡枝子沒食子酸含量為7.57 mg/g。
