一種改進的基因功能網(wǎng)絡(luò)術(shù)語相似度計算方法*
2019-03-27 09:38:32劉志明羅凌云歐陽純萍萬亞平
醫(yī)學(xué)信息學(xué)雜志 2019年2期
唐 智 劉志明 羅凌云 歐陽純萍 萬亞平
(南華大學(xué)計算機學(xué)院 衡陽 421001)
1 引言
基因本體(Gene Ontology,GO)是生物醫(yī)學(xué)領(lǐng)域最成功的本體之一,為描述基因(基因產(chǎn)物)的分子功能、生物過程等相關(guān)信息提供一個規(guī)范、準(zhǔn)確的術(shù)語集,目前被廣泛應(yīng)用于生物醫(yī)學(xué)相關(guān)研究領(lǐng)域[1]。主要體現(xiàn)在基因功能比較與分析、蛋白質(zhì)相互作用預(yù)測、基因集合富集分析等諸多領(lǐng)域,由此成為不可或缺的生物醫(yī)學(xué)本體。基因本體最初由基因本體組織(Gene Ontology Consortium)于1998年建立[2]。隨著基因本體的發(fā)展,越來越多的模式生物數(shù)據(jù)庫加入基因本體組織中,包括大多數(shù)主要的植物、動物和微生物數(shù)據(jù)庫,截至目前基因本體已經(jīng)能夠為100多個物種提供注釋信息[3]。
在生物學(xué)中,從特定的功能信息出發(fā),查找與其功能相似或者相關(guān)的蛋白質(zhì),對這些蛋白質(zhì)間關(guān)聯(lián)程度進行比較量化是生物信息學(xué)研究中經(jīng)常遇到的問題[4]。然而功能相似或者相關(guān)的蛋白質(zhì)并不一定具有較強的序列上的相似性,基因本體的出現(xiàn)為解決這一問題提供新的途徑。研究表明如果兩個基因產(chǎn)物的功能相似,那么它們在GO中注釋的術(shù)語就越相近[5]。所以GO應(yīng)用的一個重要方面就是對術(shù)語語義的相似性進行度量,從而計算基因產(chǎn)物的相似度[6]。為更好地計算術(shù)語的相似度,本文提出一種基于融合高斯核函數(shù)的重啟隨機游走的基因本體術(shù)語相似度算法RWRSM。
2 現(xiàn)有基因術(shù)語間語義相似度計算方法
2.1 基于基因本體的方法
近年來高通量生物學(xué)技術(shù)的顯著改進導(dǎo)致生物學(xué)數(shù)據(jù)的指數(shù)增加。基因本體是用于解釋生物實驗結(jié)果的最流行的生物信息學(xué)資源之一。GO提供結(jié)構(gòu)化、受控制的術(shù)語詞匯表,通過分子功能、生物過程和細胞成分3種屬性來描述基因[7]。在每個類別中術(shù)語被構(gòu)造為有向無環(huán)圖(Directed Acyclic Graph,DAG)。基于GO的語義相似性已成功應(yīng)用于許多研究領(lǐng)域,如基因功能預(yù)測[8]、基因網(wǎng)絡(luò)分析[9-10]、同源性分析[11]、基因關(guān)聯(lián)可視化[12]和缺失價值估算[13-14]。基于基因本體來計算基因功能相似性的方法[15-20]可分為4種類型:基于路徑長度的方法、基于節(jié)點信息的方法、綜合方法和基于基因功能網(wǎng)絡(luò)的方法。基于路徑長度的方法通過考慮GO拓撲結(jié)構(gòu)信息[21]來計算相似度。最近提出的方法稱為相對特異性相似性(Relative Specificity Similarity,RSS),考慮兩種類型的長度信息:從給定術(shù)語對到其最接近的葉子術(shù)語的邊長和到最低共同祖先(Lowest Common Ancestor,LCA)節(jié)點的邊長關(guān)系。但是基于邊緣的方法完全依賴于基因本體的拓撲結(jié)構(gòu)。這種方法不能在相同的拓撲水平上區(qū)分術(shù)語。對于基于節(jié)點的方法,這些方法依賴于特定的分類法。該方法利用信息量最大的共同祖先(Most Informative Common Ancestor,MICA)的信息內(nèi)容(Information Content,IC)來測量兩個GO術(shù)語之間的相似性[22]。評估測試顯示結(jié)果與蛋白質(zhì)序列相似性一致,但是基于節(jié)點的方法僅考慮注釋,忽略GO的拓撲信息。在綜合組中,提出在GO中使用更多信息的方法。混合相對特異性相似度(Hybrid Relative Specificity Similarity,HRSS)使用4種類型的信息(信息內(nèi)容、結(jié)構(gòu)拓撲、注釋和MICA)來計算語義相似性[21]。InteGO方法提出一種基于秩的方法來整合多種現(xiàn)有相似性方法,稱為種子方法,以考慮GO的更多方面[16]。InteGO2方法通過投票方法從一組方法中選擇最合適的方法,并基于元啟發(fā)式搜索方法[12]整合這些選定的方法。評估測試表明綜合方法比種子方法表現(xiàn)更好。但是所有這些方法都只基于GO結(jié)構(gòu)信息,忽略GO的不準(zhǔn)確表示和缺失的信息等問題。如只有37%的擬南芥基因具有GO的所有3個結(jié)構(gòu)域的實驗注釋[23]。因此GO中的不完整信息可能導(dǎo)致低質(zhì)量的相似性。
2.2 基于網(wǎng)絡(luò)的方法
最近提出一種基于網(wǎng)絡(luò)的計算方法(Network-based Similarity Measure,NETSIM),通過整合基因關(guān)聯(lián)和GO拓撲結(jié)構(gòu)和注釋來解決這些問題[18]。基于代謝反應(yīng)圖的實驗表明通過結(jié)合基因關(guān)聯(lián)可以增強語義相似性,但NETSIM只考慮網(wǎng)絡(luò)中的直接鏈路,使計算過程中只使用基因協(xié)同功能網(wǎng)絡(luò)中的部分信息,實際上除直接連接的基因?qū)ν膺€應(yīng)考慮基因功能網(wǎng)絡(luò)中包含的間接基因相互作用。因此一種新的基于網(wǎng)絡(luò)的NETSIM2被提出,通過隨機游走的方法考慮基因協(xié)同網(wǎng)絡(luò)中的直接和間接相互作用,代謝反應(yīng)圖的實驗表明結(jié)果遠高于之前所有算法,但是NETSIM2方法測量出的LFC值不夠穩(wěn)定,大部分LFC值很小。為解決這個問題本文通過使用高斯核函數(shù)來計算邊權(quán)重,生成歸一化加權(quán)矩陣PT,通過隨機游走算法得到每對基因之間的相關(guān)性得分。最后基于NETSIM2原有算法計算術(shù)語對相似度。與之前方法相比本文算法融合高斯核函數(shù)與隨機游走計算基因相關(guān)性得分,以考慮直接和間接的交互,通過有效地整合基因功能網(wǎng)絡(luò)大大提高LFC值的穩(wěn)定性。
3 改進的基因術(shù)語間語義相似度計算方法
3.1 融合高斯核函數(shù)與隨機游走計算基因相關(guān)性得分
3.1.1 生成隨機游走圖 重啟隨機游走圖構(gòu)建基本思路是: 將訓(xùn)練集合D中的每個訓(xùn)練數(shù)據(jù)d∈D映射為圖中的一個點, 根據(jù)兩點之間的相關(guān)程度決定是否用邊將兩點相連。為方便求解, 一般構(gòu)建成完全圖, 將兩點之間的相似度作為邊的權(quán)重。當(dāng)兩點之間的相似度很小或者為0時, 節(jié)點之間邊的權(quán)重做零值處理。假設(shè)加權(quán)圖用G=(V,E)表示,V是圖G中的節(jié)點集合,E是圖G中邊的集合, 其中節(jié)點個數(shù)為n。邊權(quán)重使用高斯核函數(shù)計算, 公式如下:
(1)
其中σ是個參數(shù)變量,經(jīng)驗設(shè)定σ=∑i≠i||xi-yi||/[n(n-1)]。dij為兩個圖像節(jié)點之間的歐式距離,表示為:
(2)
3.1.2 隨機游走過程 在使用重啟隨機游走算法之前先根據(jù)權(quán)重矩陣計算圖G的概率轉(zhuǎn)移矩陣P:
(3)
通過以下步驟計算基因之間的相關(guān)性得分。首先根據(jù)計算出的概率轉(zhuǎn)移矩陣P,然后生成歸一化加權(quán)矩陣PT。最后基于RWR的方法可以描述如下:
Di(t+1)=a·PTDi(t)+(1-a)ei
(4)
其中ei是表示初始狀態(tài)。公式(4)的穩(wěn)態(tài)解為:
Di=(1-a)(1-aPT)-1ei
(5)
其中Di是|V|×1向量,ei是|V|×1起始向量。(1-a)被定義為重啟概率,其值在0和1之間。之后可以得到矩陣S,保存N(V,E)中每對基因之間的相關(guān)性得分。
3.2 計算兩個GO術(shù)語之間的相似性
根據(jù)Peng等在以往的工作[24]中所表示的方法,計算兩個術(shù)語之間的相似性,這兩個基因本體術(shù)語對結(jié)合來自基因功能網(wǎng)絡(luò)和GO的信息。設(shè)t1和t2為兩個術(shù)語。將D(t1,t2)定義為基因集距離,以計算由t1和t2注釋的基因組之間的相似性。D(t1,t2)定義為:
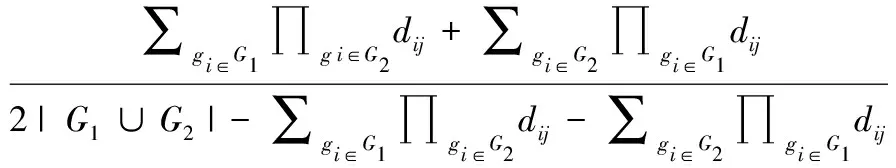
(6)
其中G1和G2分別是由t1和t2注釋的基因集合。dij是兩個基因之間的距離得分,dij=1-Sij。Sij是基于RWR的方法計算基因i和j之間的相關(guān)性得分。所有術(shù)語對的基因集距離在0~1之間歸一化。然后基于標(biāo)記為U的路徑約束注釋來計算兩個術(shù)語之間的相似性。在傳統(tǒng)的基于LCA的方法中,考慮LCA的所有后代。路徑約束注釋方法僅使用與比較術(shù)語最相關(guān)的術(shù)語。相關(guān)術(shù)語集包括3個部分:由術(shù)語t1和t2注釋的基因集、由共同父節(jié)點注釋的基因集p及其后代項在t1或t2到p的路徑上。設(shè)t1和t2為兩個GO的兩個術(shù)語對,p為它們的共同祖先。t1和t2之間的相似性根據(jù)Peng等在以往工作[24]中提出的方程定義。
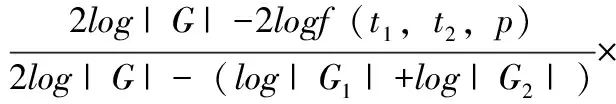
(7)
其中Gp(或G)是由共同祖先術(shù)語p(或根詞)及其后代注釋的基因集。在等式中f(t1,t2,p)基于路徑約束注釋計算相似性,定義如下:
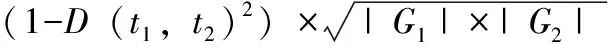
(8)
h(t1,t2)是測量共同父母的特異性,定義如下:
h(t1,t2)=D(t1,t2)2×|G|+(1-D(ti,t2)2)×
max(|G1|,|G2|)
(9)
在公式(9)中,左側(cè)部分測量從項t1和t2到p的距離,右側(cè)部分計算從p到根的距離。如果存在多于一個最低共同祖先則選擇最高得分作為t1和t2之間的相似性。
4 術(shù)語相似度算法性能測試與分析
4.1 基于基因相似度的模型測試方法
雖然有很多方法可以評價基因功能相似度,但是沒有直接的方法來衡量兩個基因本體術(shù)語的相似度。因此首先基于術(shù)語相似度計算得到基因相似度,然后借助基因之間關(guān)系來衡量術(shù)語相似度計算模型性能。這一方法在相關(guān)研究中被廣泛使用。為保證公平性,對于所有被比較的模型,通過使用相同的基因本體術(shù)語相似度來計算基因相似度并用此結(jié)果衡量術(shù)語相似度計算模型性能。如果計算某一物種對應(yīng)的所有基因本體術(shù)語之間的相似度,就可以計算該物種中任意兩個至少被一個基因本體術(shù)語注釋的基因之間的相似度。給定兩個基因gi和gj以及注釋它們的術(shù)語集合Ti和Tj,為計算基因相似度,利用Wang等提出的方法將多對術(shù)語相似度累積成基因相似度,如公式(10)所示:
(10)
公式中對于屬于集合Ti的每個術(shù)語,sim(t,Ti)表示術(shù)語t和術(shù)語集合Ti中術(shù)語之間相似度的最大值,如公式(11)所示:
sim(t,Tj)=,maxtj∈TjS(t,Tj)
(11)
公式中S(t,t1)表示t和tj的所有最小公共祖先中S(t,ti,p)的最大值。
基于EC編號(酶學(xué)委員會)組信息進行評估,由相同EC編號標(biāo)記的基因具有相似功能。基因根據(jù)其EC編號(完整的4位數(shù))分組到不同類別。然后測試同一類別的基因是否具有更高的相似性。在數(shù)學(xué)上使用記錄的倍數(shù)變化(Logged Fold Change,LFC)度量[24]進行定量評估。對EC編號的ei類別LFC分數(shù)計算如下:
(12)
其中G(ei)是基因組,是ei由標(biāo)記類別的基因組成;其中滿足G(ei)∩G(ei)=?,diffa(ei,ej)定義如下:
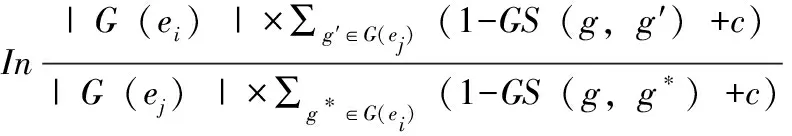
(13)
其中G(ei)是不包括g的ei基因集;c是拉普拉斯平滑參數(shù);GS(g,g′)、GS(g,g*)由公式(10)定義。等式(13)測量EC間距離和EC內(nèi)部距離之間的差異。基于公式 (12)中對對數(shù)差異倍數(shù)(LFC)得分的定義,如果一個模型的對數(shù)差異倍數(shù)得分越高,那么該模型的性能越好。
4.2 實驗數(shù)據(jù)與結(jié)果
4.2.1 實驗數(shù)據(jù) 2018年6月從GO網(wǎng)站下載GO結(jié)構(gòu)和注釋(www.geneontology.org/)工作中只使用“is-a”、“part-of”兩種術(shù)語之間的語義關(guān)系。考慮到數(shù)據(jù)量太大、時間復(fù)雜度過高,所以使用基因數(shù)目相對較少的YeastNet中包含的基因關(guān)聯(lián)對酵母菌進行評估測試。酵母菌的EC分組可以從www.yeastgenome.org/下載。
4.2.2 實驗結(jié)果 通過比較不同EC類別和相同類別的基因之間的關(guān)系,根據(jù)GO的相似性來評估本文提出的改進算法性能。使用YeastNet中包含的基因關(guān)聯(lián)對酵母進行評估測試。LFC評分作為標(biāo)準(zhǔn)與目前該領(lǐng)域LFC評分最高的兩種算法NETSIM和NETSIM2進行比較。NETSIM、NETSIM2與本文中改進的算法RWRSM測量比較每個EC組的LFC得分,結(jié)果顯示本文算法在所有104組EC編碼中有88組具有最高LFC得分,占所有分組的84.6%,而其他算法只有15個EC中具有最高LFC得分,基于NETSIM、NETSIM2及算法RWRSM對酵母菌數(shù)據(jù)表現(xiàn)最佳LFC評分的EC數(shù)量,見圖1。RWRSM算法的LFC評分針對與酵母菌的EC分組結(jié)果顯示在所有評估測量中RWRSM的平均值最高,見圖2。3種計算方法計算出的LFC平均值分別是RWRSM 0.575、NETSIM 20.547、NETSIM 0.534。3種算法的折線統(tǒng)計,見圖3。其中RWRSM算法的折線趨于穩(wěn)定在0.6左右,并且在所有EC類別中最高LFC分值占比達到85%以上,明顯優(yōu)于其他算法。
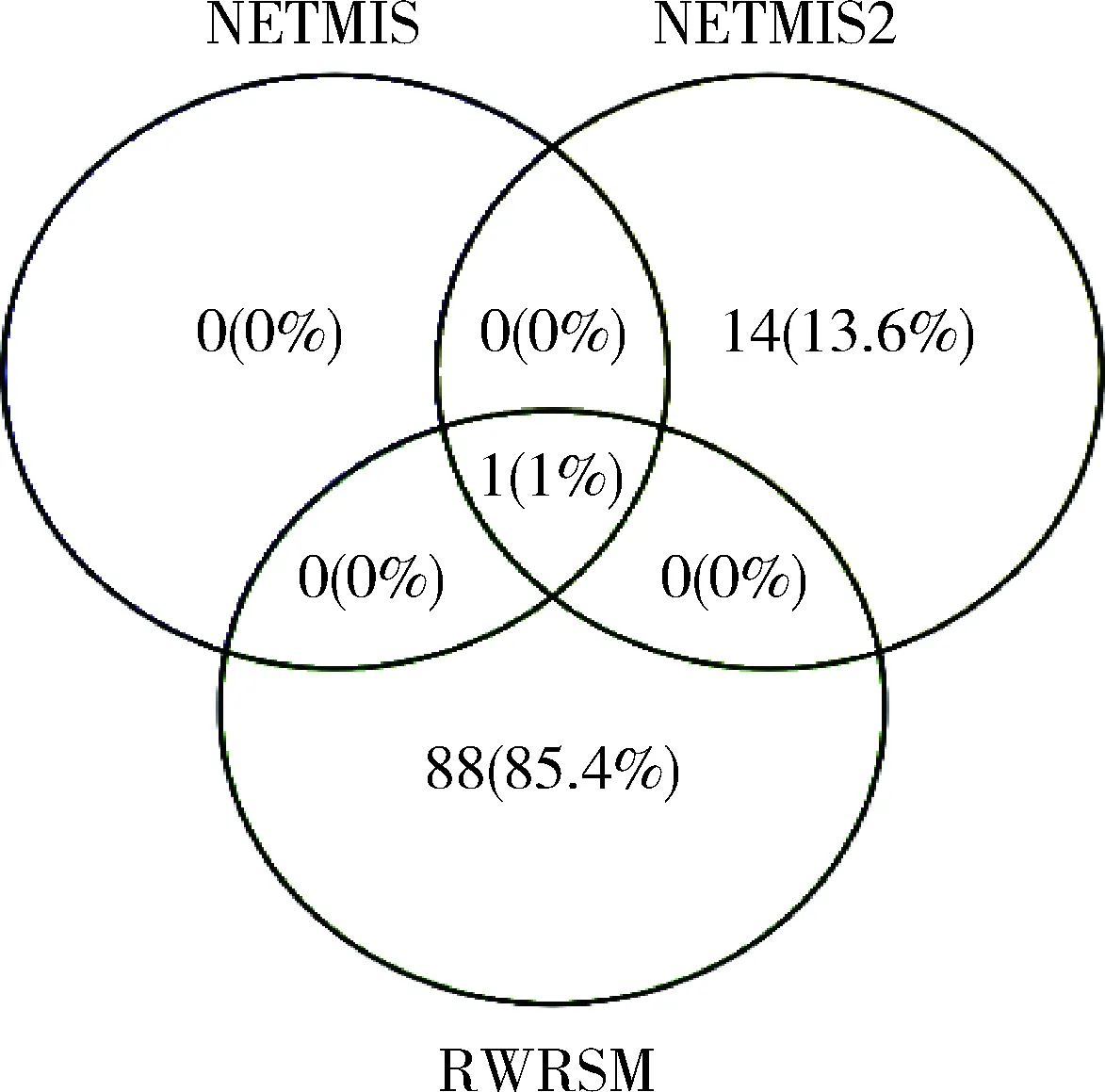
圖1 基于NETSIM、NETSIM2及RWRSM算法在酵母菌中表現(xiàn)最佳LFC評分的EC數(shù)量
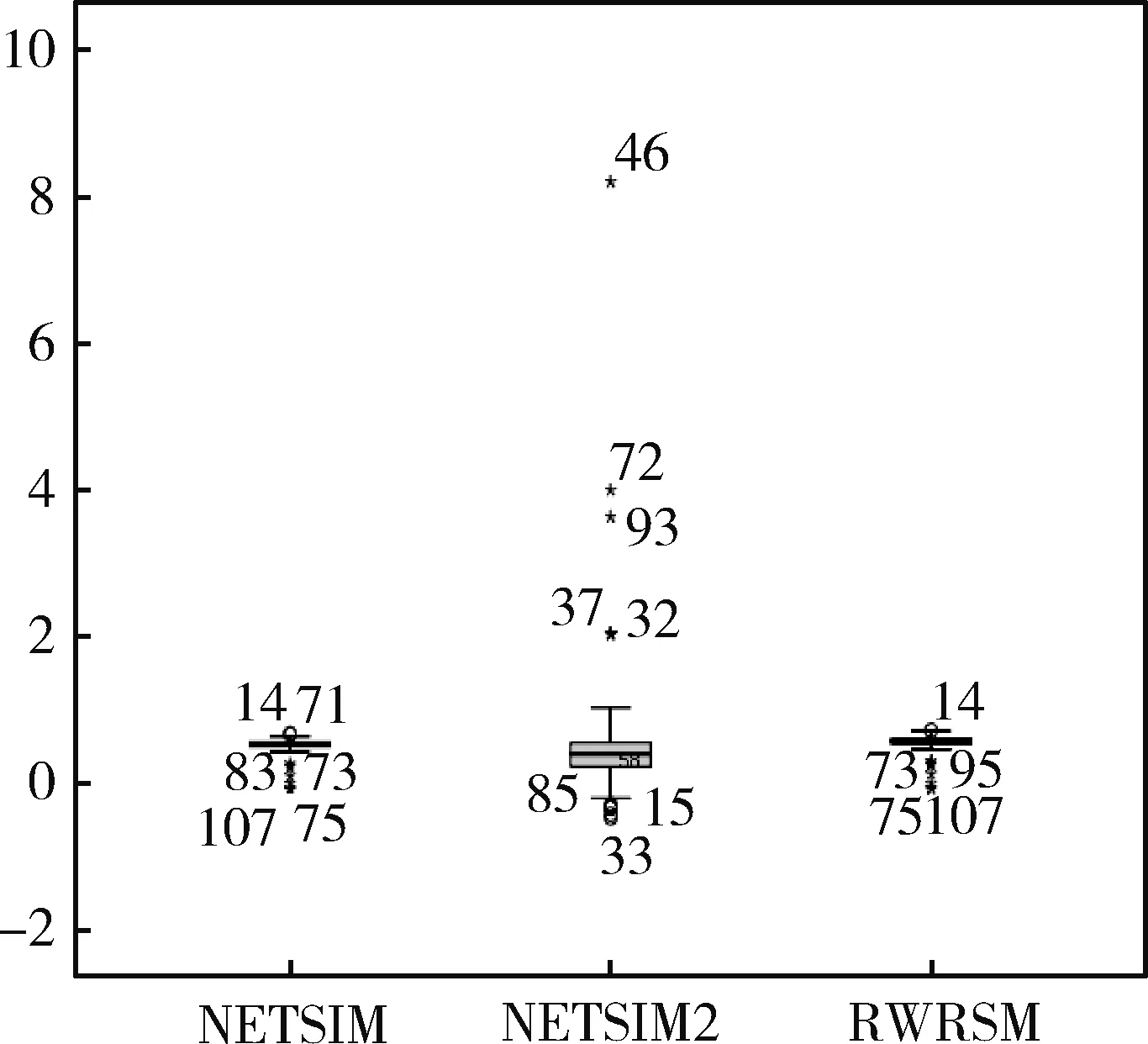
圖2 GO酵母LFC相似性測量得分性能比較
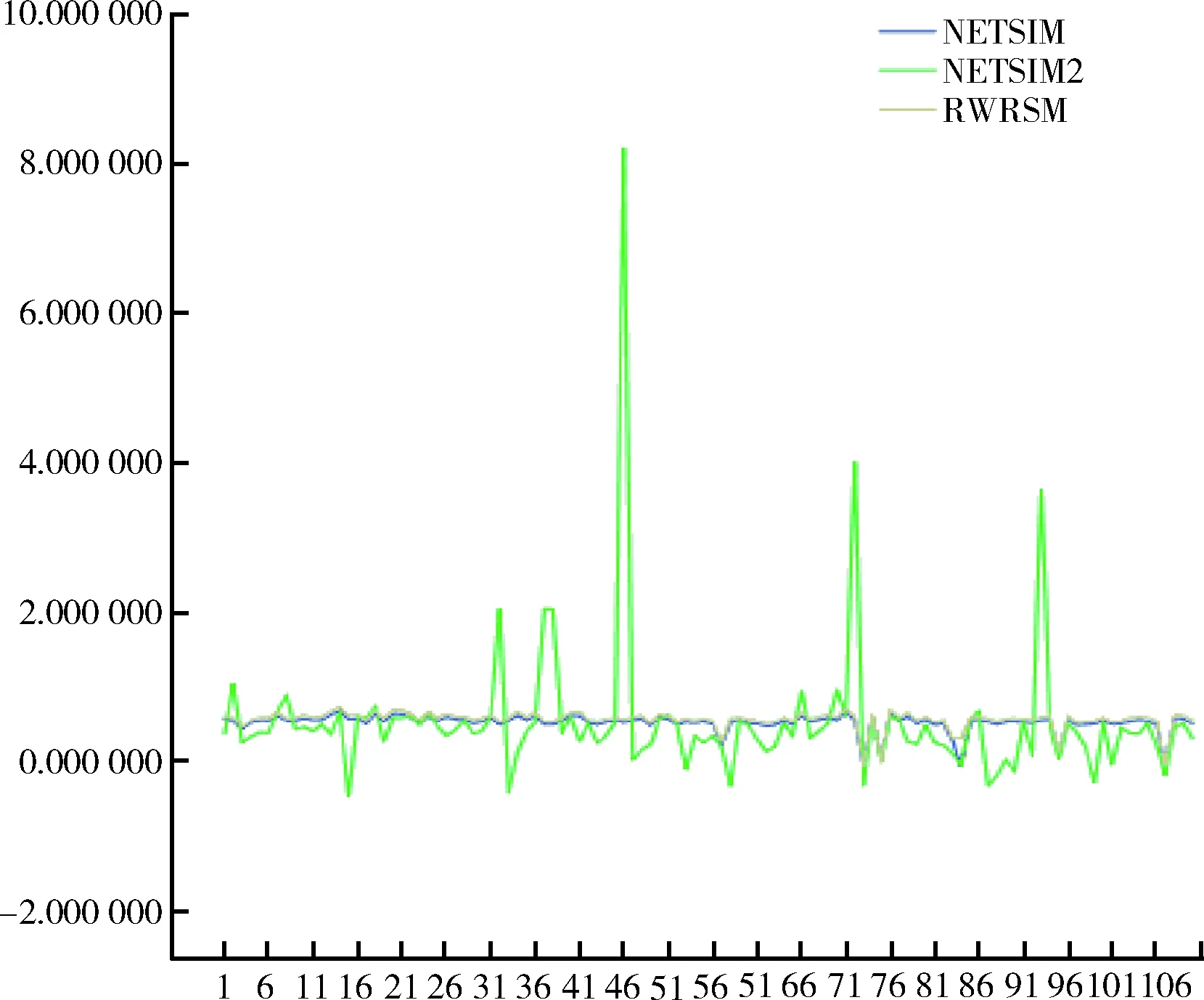
圖3 3種算法LFC相似性得分性能折線
5 結(jié)語
基因本體論是用于描述基因和基因產(chǎn)物特性的最流行的生物信息學(xué)資源之一。計算基于GO的基因功能相似性已被廣泛用于多個研究領(lǐng)域。然而低質(zhì)量的相似性可能源于GO的不完整信息和有限的注釋量。NETSIM通過考慮基因關(guān)聯(lián)、GO結(jié)構(gòu)和注釋來解決這些問題。但其僅使用基因共功能網(wǎng)絡(luò)中的本地關(guān)聯(lián)信息,因為NETSIM僅考慮網(wǎng)絡(luò)中的直接鏈路。后來提出的NETSIM2雖然考率到網(wǎng)絡(luò)結(jié)構(gòu)的全局性,但是實驗結(jié)果表明LFC評分不夠穩(wěn)定。本文提出一種新的基于NETSIM2網(wǎng)絡(luò)的改進算法,對基于RWR的方法考慮共功能網(wǎng)絡(luò)全局結(jié)構(gòu),融合高斯核函數(shù)得到權(quán)重矩陣,包括3個步驟:首先,給定基因共功能網(wǎng)絡(luò)融合高斯核函數(shù)得到權(quán)重矩陣,基于隨機游走和重啟方法計算兩個基因之間的相關(guān)性得分矩陣;其次,通過組合來自共功能網(wǎng)絡(luò)和GO的信息計算兩個GO項之間的相似性;最后,選擇GO術(shù)語對使用基于標(biāo)準(zhǔn)分數(shù)的方法測量兩個基因的相似性。EC實驗結(jié)果表明本文算法在酵母數(shù)據(jù)集的所有測量中表現(xiàn)最佳,LFC結(jié)果更加穩(wěn)定。在所有104個EC中有88個具有最高LFC得分,占所有分組的84.6%,而其他算法只有15個EC中具有最高LFC得分,平均值也明顯高于其他算法,測量出的LFC評分結(jié)果更加穩(wěn)定。
猜你喜歡
兒童故事畫報(2019年5期)2019-05-26 14:26:14
中華手工(2017年2期)2017-06-06 23:00:31
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
中外會展(2014年4期)2014-11-27 07:46:46
建筑創(chuàng)作(2001年3期)2001-08-22 18:48:14
祝您健康(1987年3期)1987-12-30 09:52:32
祝您健康(1987年2期)1987-12-30 09:52:28

- 醫(yī)學(xué)信息學(xué)雜志的其它文章
- 依托學(xué)科競賽的醫(yī)藥信息人才創(chuàng)新能力和工程素質(zhì)培養(yǎng)研究*
- 多來源作者數(shù)據(jù)加工策略與實現(xiàn)
——以西太平洋地區(qū)醫(yī)學(xué)索引為例* - 基于慕課和小規(guī)模私密在線課程的移動學(xué)習(xí)在醫(yī)學(xué)計算機教學(xué)中的應(yīng)用研究*
- 基于三級等保標(biāo)準(zhǔn)的醫(yī)院信息安全體系建設(shè)實踐
- 深度學(xué)習(xí)基礎(chǔ)上的中醫(yī)實體抽取方法研究*
- 基于本體語義增強和多源數(shù)據(jù)融合的石墨烯醫(yī)學(xué)應(yīng)用前沿探測*