誤診為IHH的單純男性化型男性21羥化酶缺陷癥一例
2019-04-03 01:55:50劉艷霞栗夏蓮
鄭州大學學報(醫學版) 2019年2期
劉艷霞,高 月,栗夏蓮
鄭州大學第一附屬醫院內分泌科;河南省內分泌代謝中心 鄭州 450052
21羥化酶缺陷癥是由于21羥化酶缺陷導致腎上腺皮質激素合成障礙的最常見的先天性腎上腺皮質增生癥。成年男性21羥化酶缺陷癥患者癥狀往往不典型,多因不育或腎上腺腫物就診。腎上腺來源的過量雄激素及中間代謝產物的蓄積可長期抑制下丘腦-垂體-睪丸,致使黃體生成素(LH)和促卵泡刺激素(FSH)水平下降,睪丸不能發育成熟,易被誤診為低促性腺激素性性腺功能減退癥(isolatedhypogonadotropichypogonadism,IHH),導致誤診誤治。現報道一例誤診為IHH的21羥化酶缺陷癥,以期提高對這兩種疾病的認識和診斷水平。
1 病例資料
1.1一般資料患者,男,30歲,因婚后未避孕不育8 a,至外院查性激素六項: LH 0.007 mIU/L(正常參考值1.000~12.500 mIU/L,后文括號內均為正常參考值), FSH 0.012 mIU/L (1.000~12.000 mIU/L), 雌二醇(E2) 135 ng/L(<75 ng/L), 泌乳素(PRL) 137.9 mIU/L (42.5~414.0 mIU/L), 睪酮(T)3.5 μg/L(2.8~12.0 μg/L),孕酮(P)14.7 μg/L(0.1~2.0 μg/L)。睪丸超聲示:左側睪丸22 mm×14 mm×14 mm,右側24 mm×14 mm×17 mm,輪廓清晰,包膜光滑。垂體MRI示:垂體前葉下部片狀稍長T2信號,增強掃描未見明顯異常。精液分析:排精量4.6 mL(>2 mL),酸堿度pH7.4 (pH7.2~8.0),精子活動率0%(>60%), 精子活力不正常,精子密度 0×106個/mL(>20×106個/mL),精液離心鏡檢未找到精子。睪丸穿刺活檢:穿刺液0.7 mL,高倍鏡下未見精子。達比佳激發試驗(靜脈注射戈那瑞林60 min)LH 1.359 IU/L。肝功能、腎功能、電解質均正常。診斷為特發性IHH,擬行促性腺激素替代治療。
經我院會診,認為患者LH與FSH水平低,T水平正常,未服用任何藥物,與IHH臨床特點及體征不相符,考慮睪酮為腎上腺來源。進一步完善病史:患者性生活可正常完成,每周2~3次;體格檢查示嗅覺正常,有胡須,身高160 cm,體重64 kg,體重指數25.0 kg/m2,血壓120/85 mmHg(1 mmHg=0.133 kPa),陰毛濃密呈菱形分布、色深、未到臍,陰莖長度約9 cm,雙側睪丸約4 mL(Prader睪丸計)。追蹤生長發育史:足月順產,頭先露,出身后無拒食、嘔吐,6~8歲時身高較同齡人高,9歲時陰莖有不自主勃起現象,未就診。父母已故,愛人體健,父母及兩個妹妹均體健,兩妹自然受孕。既往史、個人史無特殊。腎上腺CT(圖1)示雙側腎上腺增粗,見等密度結節影,邊緣不整,密度均勻。
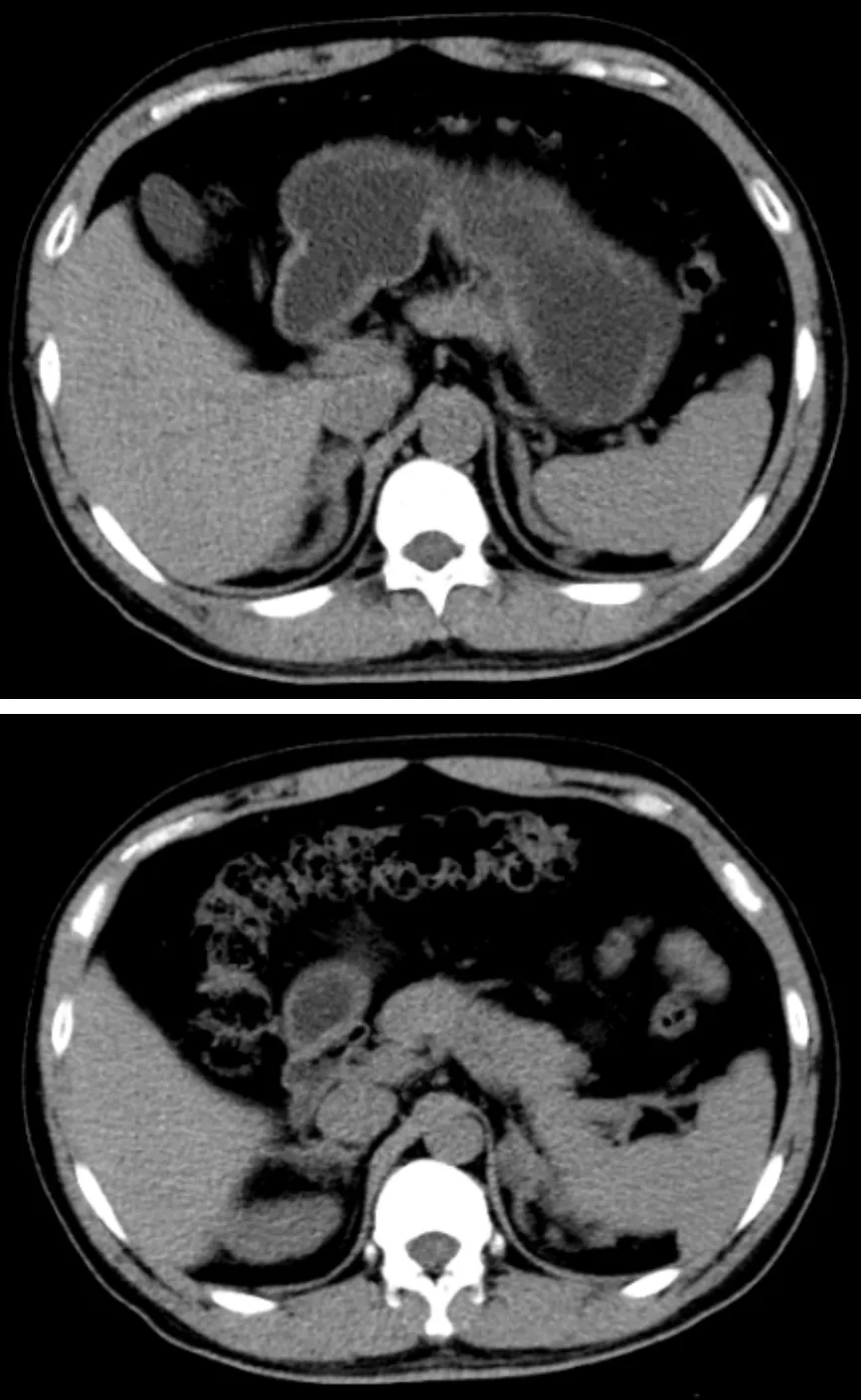
圖1 患者腎上腺CT平掃表現
1.2實驗室檢查我院采用全自動生化儀測定肝腎功能、血電解質等,化學發光法測定LH、FSH、T、硫酸脫氫表雄酮(DHEAS)、雄烯二酮(AD)、腎上腺皮質激素(ACTH)、皮質醇(COR)等內分泌激素。性激素六項:FSH為0.16 mIU/L(0.95~11.95 mIU/L),LH為0.01 mIU/L(1.14~8.75 mIU/L), P為5.74 μg/L(<0.10~0.20 μg/L),E2為31 ng/L(11~44 ng/L),T為4.98 μg/L(1.42~9.23 μg/L),PRL為14.18 μg/L(5.18~26.53 μg/L);其他內分泌相關激素: DHEAS為376 μg/dL(80~560 μg/dL,1 μg/dL=0.01 mg/L),AD>10.0 μg/L(0.6~3.1 μg/L),17-羥孕酮(17-OHP)為47.88 μg/L(0.61~3.34 μg/L);腎素活性臥位為1.18 μg/(L·h)[(0.15~2.33)μg/(L·h)],立位為5.54 μg/(L·h)[0.1~6.56 μg/(L·h)];醛固酮臥位為243.1 ng/L(30.0~160.0 ng/L),立位為243.1 ng/L(30.0~160.0 ng/L)。ACTH-COR節律見表1。

表1 ACTH-COR節律
1.3基因檢測獲得患者知情同意后,抽取患者靜脈血5 mL,EDTA抗凝,應用過柱法檢測試劑盒(美國AIAGEN公司)抽取外周血DNA,送至上海韋翰斯生物醫藥科技有限公司用ABI3700熒光自動測序儀測序,用Primer 5.0設計特異性引物,結果與NCBI網站的CYP21基因序列(Gene ID為1589)進行比對。Sanger測序結果(圖2)顯示,患者CYP21A2基因第6外顯子存在雜合錯義突變c.518T>A(p.I173N)。
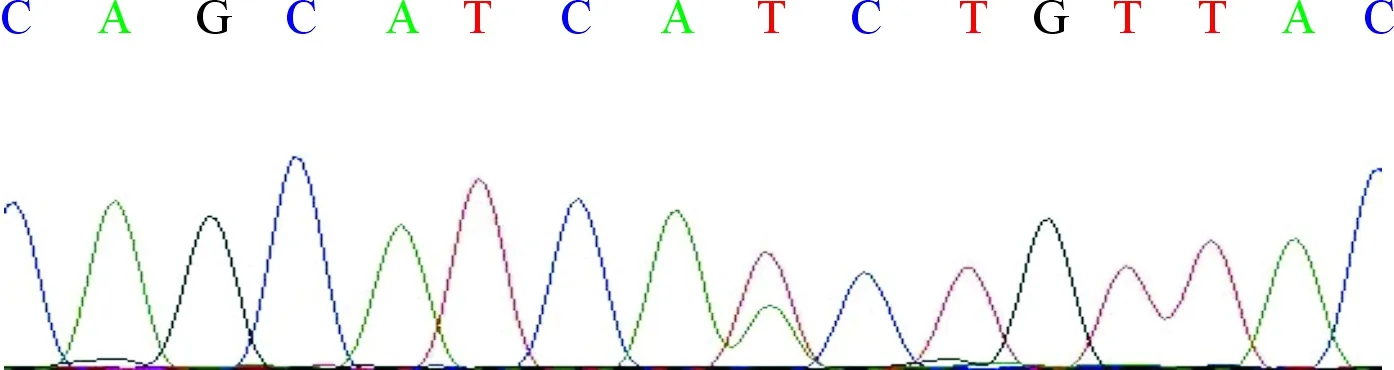
圖2 患者Sanger測序峰圖
2 討論
該例患者為30歲男性,婚后不育8 a,性生活正常(未使用藥物),內分泌檢查提示LH、FSH低,睪酮正常,達必佳試驗LH可以上升到1.359 IU/L,故外院診斷為IHH。但患者身材偏矮,陰毛濃密呈菱形分布,陰莖長度正常,實驗室檢查睪酮正常,考慮腎上腺為睪酮的另一個來源,故進一步評估腎上腺相關激素,結果顯示DHEAS、AD、17-OHP和ACTH均升高,基因檢測發現CYP21A2基因第6外顯子存在雜合錯義突變:c.518T>A(p.I173N),故最終診斷為單純男性化型21羥化酶缺陷癥。
21羥化酶缺陷癥是由于在腎上腺皮質激素合成途徑中21羥化酶缺陷導致的常染色體隱性遺傳病,是先天性腎上腺皮質增生癥最常見的類型,根據酶缺乏程度分為失鹽型、非經典型和單純男性化型[1]。失鹽型患者21羥化酶完全缺乏,皮質醇和皮質酮合成均出現障礙,胎兒期起病,可出現高鉀血癥、低鈉血癥、代謝性酸中毒、低血糖癥等代謝紊亂癥狀,甚至腎上腺危象。非失鹽型患者由于癥狀不典型,就診率低。成年男性21羥化酶缺陷癥患者生育能力下降,常因婚后不育及睪丸腫物而就診[2],臨床極易誤診。
特發性IHH是臨床上造成男性性發育異常的疾病之一,典型表現為小陰莖、睪丸體積小(1~3 mL)、第二性征不發育或發育不全,骨骺閉合延遲呈現“宦官樣”體征,促性腺激素水平低或正常,且睪酮水平≤3.47 nmol/L[3]。單純男性化型21羥化酶缺陷癥患者21-羥化酶不完全缺乏,醛固酮合成正常,雄激素過量,男性可表現為外周性性早熟,陰毛提早出現,陰莖增大且容易勃起,但睪丸體積仍為青春發育前大小,體格發育過快,骨齡提前,成年后身材偏矮,皮膚黏膜色素沉著,腎上腺來源的睪酮可使第二性征發育,維持性生活。所以詳細的病史采集和體格檢查是鑒別二者的重要依據之一。
男性21羥化酶缺陷癥患者不育原因之一為CYP21活性降低或消失,P和17-OHP不能被轉化為11-去氧皮質酮,COR合成減少,對下丘腦和垂體的反饋抑制作用減弱,ACTH分泌增加,刺激腎上腺皮質(主要為束狀帶)增生,導致過量17-OHP與P產生。過量的17-OHP和P一部分在17,20-碳裂解酶作用下進入雄激素合成途徑。過量的雄激素可在外周通過芳香化酶的作用部分轉化為雌激素,過量的雄激素、雌激素及17-OHP、P、AD等中間代謝產物共同長期抑制下丘腦-垂體-睪丸,反饋抑制LH和FSH;睪丸不能發育成熟,表現為睪丸細小、堅硬,前列腺發育不良,曲細精管和睪丸間質細胞發育異常、精子生成障礙,從而導致不育[4]。因腎上腺雄激素的作用,男性第二性征存在,陰莖正常或增大,有時甚至表現為毛發增多,但患者身材偏矮,多伴有中心性肥胖、高血壓、胰島素抵抗甚至2型糖尿病等代謝綜合征的表現,也影響患者生育能力[5]。睪丸腎上腺殘余瘤是導致成年男性21羥化酶缺陷癥不育的另一重要因素。增高的ACTH和血管緊張素Ⅱ(AT-Ⅱ)刺激睪丸內腎上腺皮質殘余細胞增生,從而發生睪丸腎上腺殘余瘤,壓迫睪丸網導致曲細精管梗阻,其分泌的腎上腺類固醇類激素對睪丸間質細胞和生殖細胞有生殖毒性,嚴重者可致不可逆的睪丸功能損害[6]。Bouvattier等[7]統計了2011至2014年間219例男性非經典型21羥缺陷癥患者,42%有少精癥或無精癥,合并腎上腺殘余瘤的患者少精和無精發生率達70%。
21羥化酶缺陷癥部分患者需終生口服腎上腺皮質激素替代治療[2],緩解COR和醛固酮分泌不足的癥狀,抑制腎上腺來源的過量雄激素;糖皮質激素強化治療可提高生育功能受損患者的生育能力[8]。國內外個案報道[9-10]顯示合并低ACTH患者的生育能力是可逆的,經短期治療后均可恢復生精能力。腎上腺皮質激素治療失敗可考慮聯合應用促性腺激素治療促進精子生成及輔助生殖技術。
綜上所述,成年男性21羥化酶缺陷癥患者癥狀不典型,應詳細詢問生長發育史、評估臨床體征,以免誤診。
