CDK12主要生理功能與其在腫瘤發生中作用的研究進展
2019-04-20 08:17:34李曉軍陶麗梅朱占弟聶元華陳敏學
生命科學研究 2019年5期
關鍵詞:研究
李曉軍,陶麗梅,朱占弟,聶元華,陳敏學,王 琛*
(蘭州大學第二醫院a.普外科四病區;b.婦產科,中國甘肅蘭州730030)
周期蛋白依賴性激酶(cyclin-dependent kinase,CDK)是調節各種細胞過程的重要激酶,可分為細胞周期相關的CDKs(如CDK1/2/4/6)及轉錄相關的CDKs(如CDK7/8/9/11/12/13)。細胞周期相關的CDKs主要通過調控機體細胞周期中各個時期的進展,直接影響細胞的增殖;轉錄相關的CDKs主要通過磷酸化C末端域的RNA結合蛋白1(RNA binding protein 1,Rbp1),調節基因轉錄(表1)。Rbp1是RNA聚合酶Ⅱ(RNA polymeraseⅡ,RNA polⅡ)的最大亞基,也是主要的轉錄調控因子。由于腫瘤細胞中廣泛存在CDK失控及Rbp1等轉錄調控因子的異常轉錄,因此Rbp1很有可能成為新的腫瘤治療靶點[1~2]。
CDK12是一種轉錄相關的CDK,可使RNA聚合酶Ⅱ碳端氨基酸(carboxy terminal domain of RNA polymerase Ⅱ,RNA pol II CTD)磷酸化,這對于DNA損傷修復(DNA damage repair,DDR)、mRNA剪接以及細胞增殖和分化至關重要[9]。研究表明,在多種惡性腫瘤(特別是乳腺癌)中廣泛存在CDK12的基因突變和過表達,抑制腫瘤中CDK12的表達,將有助于人們對CDK12具體生理功能的了解[10]。目前,CDK12抑制劑的相關研究日益受到人們關注,此類抑制劑作為部分腫瘤靶向藥物治療的前景廣闊[10~11]。本文初步探討了當前CDK12在正常細胞及某些腫瘤細胞發生、發展中的作用,這可能為存在CDK12表達異常的腫瘤的靶向藥物研究提供幫助。

表1 轉錄相關CDKs的比較Table 1 The comparison of transcription-related CDKs
1 CDK12簡介
2001年,Ko等[12]通過對HeLa細胞cDNA的研究發現了一種新的轉錄相關激酶,并將其命名為CrkRS(Cdc2-related kinase with an arginine/serinerich domain)。CrkRS由1 490個氨基酸組成,具有一個激酶結構域和脯氨酸、絲氨酸富集區,屬于典型的RS蛋白(arginine/serine-rich proteins,RS proteins)家族反式作用因子[12]。2006 年,Chen 等[13]研究發現CDK12主要結合細胞周期蛋白L1和L2,而細胞周期蛋白L1和L2位于剪接因子區,C端含精氨酸和絲氨酸富集的結構域,故CrkRS被重新命名為CDK12。CDK12主要定位于人染色體17q12-pter[14]。Moradian 等[15]通過針對果蠅 CDK12的克隆實驗發現,細胞周期蛋白K(cyclin K)為主要的CDK12結合的細胞周期蛋白。在體外,cyclin K/CDK12復合物可使RNA polⅡCTD磷酸化,即CDK12為RNA polⅡCTD致活酶[15]。與此同時,通過開展哺乳動物細胞中cyclin K與CDK12的免疫沉淀反應和質譜分析,Bla?ek等[16]得出CDK12與cyclin K的結合不但可使RNA polⅡCTD磷酸化,促進細胞轉錄,而且還與BRCA1(breast cancer susceptibility gene 1)、ATR(ataxia telangiectasia and Rad3-related)、FANC1(Fanconi anemia complementation group 1)和FANCD2(Fanconi anemia complementation group D2)等維持細胞基因穩定性和參與DDR的關鍵因子表達有關。此外,B?sken等[17]研究發現CDK12也可能作用于RNA polⅡCTD以外的底物,如轉錄因子或剪接因子等。這些研究表明CDK12可能與轉錄調控及RNA剪接等細胞過程具有相關性。然而,除RNA polⅡCTD以外,CDK12其他的磷酸化作用靶點和其相關的生物學機制仍有待闡明。
在人類基因組中,最接近CDK12的同源基因是CDK13。CDK13也被稱為CDC2L5(cell division cycle 2-like protein kinase 5),含有一個與CDK12激酶域高度序列一致性的激酶結構域(圖1)[18]。與CDK12相類似,CDK13也能使RNA polⅡCTD磷酸化,且也能結合cyclin K形成一個獨立的復合物[19],但是CDK13的作用靶點并不在RNA polⅡCTD Ser2/5。目前對于CDK13的研究遠遠少于CDK12,其功能和作用機制不甚清楚[19]。由于CDK13與CDK12的基因編碼序列相似,也有人認為這兩個激酶可能具有相似的生理功能[19]。
2 CDK12生理功能
2.1 CDK12參與轉錄調控

圖1 CDK12與CDK13的結構示意圖(參照文獻[18]修改)CDK12與CDK13都具有富含精氨酸/絲氨酸(arginine/serine-rich,RS)、富含脯氨酸(proline-rich,PRM)的結構域以及激酶結構域(kinase domain,KD),但與CDK13相比,CDK12中富含丙氨酸(alanine-rich,Ala)、絲氨酸(serine-rich,SR)結構域相對較少。Fig.1 The structure schematics of CDK12 and CDK13(revised by reference[18])CDK12 and CDK13 both have arginine/serine-rich(RS),proline-rich(PRM)domains and kinase domain(KD),but compared with CDK13,CDK12 has less alanine-rich(Ala)and serine-rich(SR)domains.
研究表明CDK12可使果蠅和人類細胞中RNA polⅡCTD的2號位絲氨酸(Ser2)磷酸化,促進細胞中的轉錄延伸,但下調CDK12活性并不影響整體轉錄速率[16~17]。Li等[20]研究發現 CDK12 是果蠅細胞中核呼吸因子2(nuclear respiratory factor 2,Nrf2)相關基因表達的關鍵酶之一,然而抑制CDK12活性,并不影響大量的DNA轉錄和mRNA翻譯。因此,CDK12可能只是促進某一組特定基因轉錄的蛋白激酶,抑制CDK12的表達在一定程度上并不影響轉錄的整體速率[20]。最近有研究指出,在短暫沉默的RNA PolⅡ釋放進入轉錄延伸階段后,CDK12可通過完全調控PolⅡ相關因子1(PolⅡ-associated factor 1,Paf1)而募集機體轉錄的基因[21];在使用特異性抗CDK12抗體進行染色質免疫共沉淀測序(chromatin immunoprecipitation sequencing,ChIP-Seq)實驗時Chirackal等[22]發現,CDK12能夠與RNA polⅡ編碼基因、啟動子以及具有轉錄活性的增強子結合,而且ChIP-Seq結果顯示CDK12與RNA polⅡ部分序列相重疊。這似乎進一步解釋了CDK12可能只促進某一組特定基因的轉錄。Johannes等[23]研發了一種CDK12抑制劑,即dinaciclib(SCH 727965),研究表明低劑量的dinaciclib(SCH 727965)會降低核心DDR基因如 BRCA1、FANCF(Fanconi anemia complementation group F)和ERCC4(excision repair cross-complementing group 4)的轉錄速率;而高劑量的dinaciclib(SCH 727965)卻只會降低超級增強子相關基因的表達,與轉錄的整體速率無明顯相關性。
CDK12調節轉錄的特異性主要與CTD中特定絲氨酸有關。之前的研究表明,在體內外CDK12均可使RNA polⅡCTD Ser2磷酸化,促進轉錄[16~17]。為了明確 CDK12 底物的性質,B?sken等[17]進行了體外激酶實驗和免疫沉淀反應實驗,結果表明CDK12可使RNA polⅡCTD Ser2和Ser5磷酸化,但此過程需要在RNA polⅡCTD Ser7輔助的基礎上進行(圖2)[24]。盡管抑制HeLa中CDK12的活性將導致細胞生長障礙,但這種改變對HeLa中RNA polⅡCTD相關絲氨酸整體磷酸化水平的影響不大[10~11]。Zhang等[11]研究表明抑制CDK12活性將導致RNA polⅡCTD Ser2磷酸化效率降低,但RNA polⅡCTD Ser5或Ser7的磷酸化卻不受影響。RNA polⅡCTD底物特異性研究一直依賴于CTD特異性抗體,如H5、H14抗體等,但這些抗體的特異性易受到CTD修飾底物的影響[25],使結果產生偏倚,故所得出的結論也容易受到質疑。Schuller等[26]研發了一種適用于修飾RNA polⅡCTD及質譜分析的細胞株,這可能為闡明CDK12及其他CDKs中單個CTD絲氨酸的特異性提供了一種有價值的途徑。
CDK12除了調控轉錄延伸外,還參與轉錄終止(圖3)[24]。裂解刺激因子77(cleavage stimulation factor 77,CstF77)是多聚腺苷酸化的重要因子之一,CDK12可通過招募CstF77使RNA polⅡCTD Ser2磷酸化,后者與MYC(myelocytomatosis oncogene)基因多聚腺苷酸化相偶聯并共同作用于mRNA 3'末端,從而終止轉錄[27]。CDK12的缺失使RNA polⅡCTD Ser2磷酸化效率減弱、裂解刺激因子64(cleavage stimulation factor 64,CstF64)減少,導致EGF(epidermal growth factor)信號激活后c-Fos(cellular oncogene fos)基因的3'端修復受損,進而影響mRNA的轉錄終止[17]。
轉錄延伸和終止是基因表達的重要調控步驟,這些過程的失調可能會改變抑癌基因或致癌基因的表達水平,從而影響腫瘤的發生發展。目前,CDK12在轉錄調控中的具體作用機制尚不完全清楚。CDK12是否會影響細胞整體轉錄或者它是否作為一種特異性的激酶作用于某一組獨特的基因(如DDR或增強子相關基因)而調控轉錄這一根本性問題仍未闡明。
2.2 CDK12參與RNA剪接
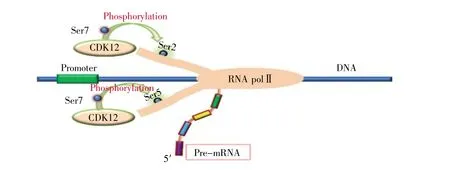
圖2 CDK12參與轉錄延伸(參照文獻[24]修改)CDK12在RNA PolⅡCTD Ser7的輔助下使RNA PolⅡCTD Ser2/Ser5磷酸化,促進轉錄延伸。Fig.2 CDK12 is involved in transcriptional extension(revised by reference[24])CDK12 phosphorylates RNA PolⅡCTD Ser2/Ser5 with the help of RNA PolⅡCTD Ser7,facilitating transcriptional elongation.
Moradian等[15]應用質譜分析確定了組成剪接體的幾個因子,包括splicing factor 2;SF2/alternative splicing factor;ASF、splicing component 35;SC35、serine/arginine rich protein 40、55、75;SRp40、SRp55和SRp75等。后續研究表明這些因子與CDK12調控的RNA剪接密切相關,但其中大多數尚未被分子內剪接測定和免疫沉淀反應所證實[10,12]。相關研究表明在果蠅神經系統發育過程中,CDK12參與了軸突蛋白IV與mRNA結合蛋白HOW的選擇性剪接[21]。另外,在RNA序列檢測實驗中,Tien等[28]通過對Her2陽性的乳腺癌細胞、三陰乳腺癌細胞、正常乳腺上皮細胞MISO進行可變剪接分析,發現CDK12可選擇性地調控3'端外顯子的可變剪接以及最后一個外顯子的可變剪接。盡管越來越多的研究表明CDK12參與RNA剪接(圖4)[24],但CDK12在RNA剪接這一過程中的具體作用及可能的作用機制仍有待研究。
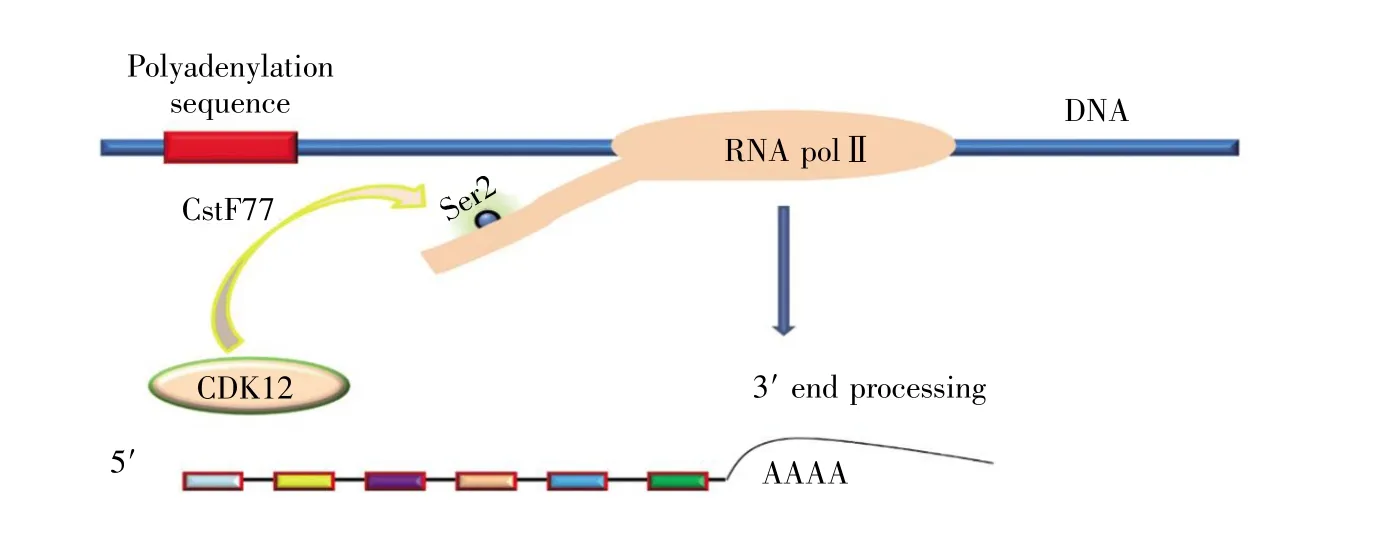
圖3 CDK12參與轉錄終止(參照文獻[24]修改)CDK12通過募集CstF77等多聚腺苷酸化因子使RNA聚合酶Ⅱ磷酸化,后者作用于mRNA 3'端,終止基因表達,促進轉錄終止。Fig.3 CDK12 is connected to the transcription termination(revised by reference[24])CDK12 phosphorylates RNA polymeraseⅡby recruiting polyadenylation factors such as CstF77,which acts on the 3'end of mRNA to block gene expression and terminate transcription.
2.3 CDK12參與細胞成熟和分化
盡管目前已經檢測到哺乳動物組織、細胞中存在CDK12的廣泛表達,但不同組織、器官中CDK12的表達水平均有差異。比如:mRNA檢測發現在人類睪丸、卵巢、白細胞和腎上腺中CDK12表達水平較高[16]。此外,在小鼠胚胎干細胞、睪丸細胞中CDK12蛋白水平普遍高于其他分化程度較高的小鼠細胞[29],這表明隨著分化程度的增高CDK12的表達水平越低。Chen等[30]研究發現CDK12和(或)CDK13的缺失可能會降低CDK5的表達,導致軸突生長減少,抑制神經元發育和分化。與之相類似的小鼠胚胎研究發現,體外培養去除CDK12或(和)cyclin K的囊胚,由于細胞凋亡增加和DNA損傷修復機制的受損,導致細胞無法大量增殖[31]。免疫沉淀與免疫印跡檢測發現CDK12在小鼠胚胎干細胞中高表達,但CDK12/cyclin K復合物的表達卻不明顯[32],且cyclin K的表達水平隨著小鼠胚胎干細胞分化程度的增高而降低,并與Oct4(octamer-binding transcription factor 4)、Sox(Sryrelated high mobility group box)和Nanog等維持細胞多能性蛋白質的表達水平相關[32]。另有研究報道,提高秀麗隱桿線蟲細胞中CDK12/cyclin K復合物的活性,可增強秀麗隱桿線蟲生殖系中RNA polⅡCTD Ser2的磷酸化水平,使其繁殖能力大幅度提高[33]。雖然,上述研究大部分證實CDK12在促進細胞的分化成熟方面扮演了極為重要的角色,但其發揮作用的可能機制尚未見報道,仍有待于進一步探索。
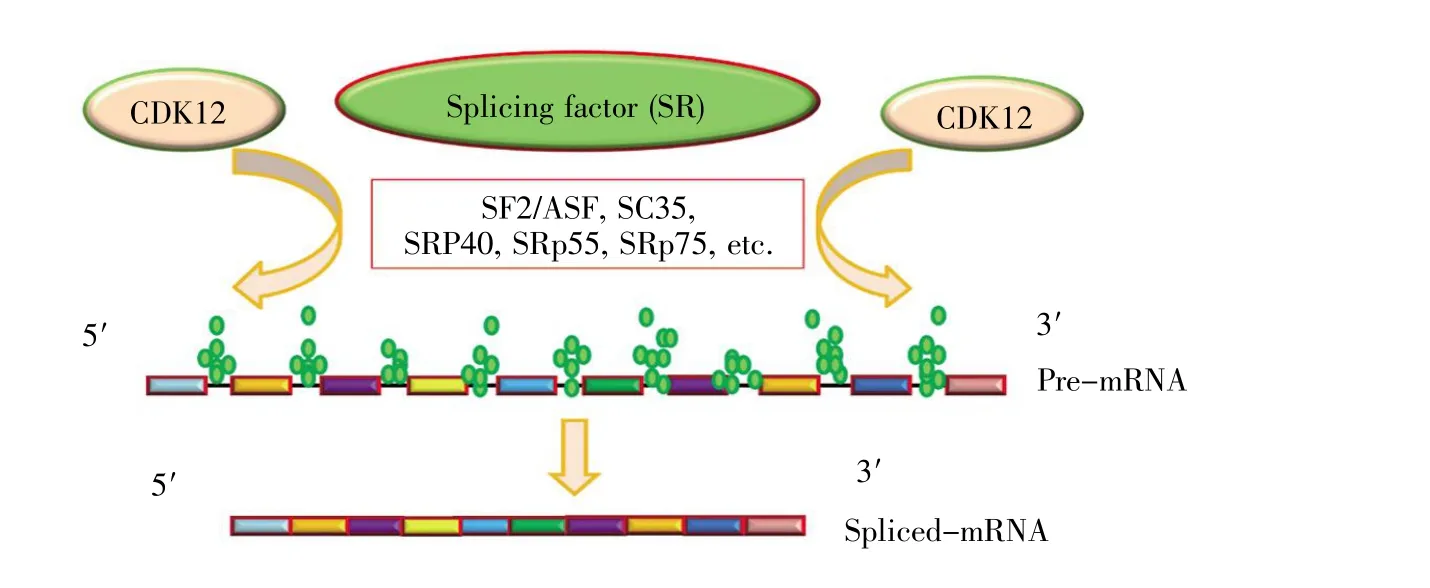
圖4 CDK12參與RNA剪接(參照文獻[24]修改)CDK12與剪接體組成因子共同作用于pre-RNA,調節pre-mRNA剪接。Fig.4 CDK12 is linked with RNA splicing(revised by reference[24])CDK12 associates with a component factor of the splicing body to act on RNA sequences and regulate pre-mRNA splicing.
2.4 CDK12參與DNA損傷修復
雖然CDK12在細胞中的具體功能及作用機制尚不完全清楚,但很明顯它在DDR中發揮著極為重要的作用[30]。研究表明,抑制CDK12使得BRCA1、ATR、FANCI、FANCD2 等維持基因穩定性關鍵因子的表達減低,同源重組(homologous recombination,HR)轉錄活性受到抑制,DNA雙鏈斷裂修復效率減低,DDR過程受阻[8,13];CDK12/cyclin K復合物的失活,一方面導致內源性DNA損傷增加,另一方面使細胞中有效執行同源重組的能力受損,進而導致DDR障礙[20,34]。盡管CDK12在DDR中的具體作用機制尚不完全清楚,但可以肯定的是CDK12在維持基因組穩定性和HR轉錄活性、促進DNA損傷修復中具有不可替代的作用。DDR受損和DNA損傷累積是癌癥的典型特征之一[35],以上結果表明CDK12缺乏與腫瘤發生發展密切相關。
3 CDK12與腫瘤的發生發展
3.1 CDK12缺失可能促進腫瘤的發生
通過對乳腺細胞中DNA損傷修復基因的研究,Bla?ek 等[16]發現 CDK12 和(或)cyclin K 的缺失導致乳腺細胞中長片段基因(>10 kb)和含外顯子數量較多的基因表達減少,DDR相關基因表達降低,包括維持基因組穩定性的關鍵調節因子BRCA1、ATR、FANCI和 FANCD2的編碼基因,CDK12/Cyc K復合物通過調節DDR基因的表達來維持基因組穩定性,CDK12的缺失可能促進乳腺細胞的突變,以及乳腺癌的發生。研究表明參與調控轉錄、剪接的因子(如CDK12)缺失,一方面直接影響哺乳動物的轉錄進程,改變大多數細胞的增殖、分化進程,進而使細胞異型性增加;另一方面導致pre-RNA異常選擇性剪接,基因序列改變,以上兩種改變大大增加了細胞癌變的概率[36~37]。總之,CDK12的缺失可能會導致基因轉錄或mRNA剪接等細胞過程失控,使細胞異型性增加,從而促進腫瘤的發生,但CDK12缺失后細胞出現異常的具體機制尚未闡明,對CDK12失活的腫瘤細胞進行質譜分析及基因檢測可能會對揭示這一規律提供幫助。
3.2 CDK12的缺失使腫瘤細胞對PARP1/2抑制劑敏感
PARP1/2(poly ADP-ribose polymerase 1/2)是DNA修復酶,是細胞凋亡核心成員半胱天冬酶(caspase)的切割底物,主要通過與DNA單、雙鏈斷裂處結合而被激活,參與調節氧化應激誘導的DDR過程[38~39]。PARP1/2抑制劑主要作用于有缺陷的同源重組,抑制腫瘤細胞DDR,發揮其抗癌活性[35]。最近研究表明在高級別漿液性卵巢癌中,CDK12的缺失使得同源重組基因受損,進而導致腫瘤細胞對PARP1/2抑制劑的敏感性增加[40]。雖然最近PARP1/2抑制劑已初步應用于臨床治療,但由于某些腫瘤對PARP1/2抑制劑產生了耐藥性,所以目前急需新的治療方案增加腫瘤對PARP1/2抑制劑的敏感性[41]。
Bajrami等[39]通過腫瘤綜合致死率的篩選發現,CDK12是增加PARP1/2抑制劑奧拉帕尼敏感性的重要因子。CDK12表達較低的卵巢癌細胞對奧拉帕尼治療更敏感,在體內異種移植實驗中奧拉帕尼對CDK12短暫失活的腫瘤細胞的治療作用得到證實[39]。Joshi等[42]也發現在CDK12失活的卵巢癌細胞系中,癌細胞對順鉑、烷基化劑美法侖和PARP1/2抑制劑奧拉帕尼的敏感性增加。此外,在Her2陽性乳腺癌細胞中下調CDK12的表達,腫瘤細胞對PARP1/2抑制劑的敏感性也明顯增強[43]。Johnson等[10]通過三陰乳腺癌及人源性乳腺腫瘤異種移植模型的研究發現,CDK12抑制劑dinaciclib可減低奧拉帕尼耐藥腫瘤細胞中受損同源重組基因的恢復,聯合應用dinaciclib及奧拉帕尼可明顯抑制腫瘤生長。因此,CDK12抑制劑的開發可作為減少腫瘤對PARP1/2抑制劑或其他DDR修復系統抑制劑耐藥性的一種治療方案。
4 展望
CDK12是一種與轉錄相關的周期蛋白依賴性激酶,對DDR、mRNA的剪接及細胞分化、成熟等多個細胞過程的運作至關重要,盡管近些年來CDK12得到了較為廣泛的研究,但我們對其功能及作用機制的了解仍然十分有限。建立裸鼠腫瘤模型,上調或沉默CDK12的表達,觀察腫瘤的生長及侵襲轉移等可能會有助于我們評估CDK12及其相關激酶在腫瘤細胞中的功能。另外,選用特異性CDK12抑制劑和腫瘤相關基因的質譜分析也將有助于鑒別CDK12究竟是一種通用的轉錄調控因子,還是某個轉錄階段的特殊因子。所有這些都可能為后期揭示CDK12在腫瘤發生發展中的具體作用機制以及腫瘤靶向治療藥物的研究提供幫助。
越來越多的研究表明腫瘤中的確存在CDK12的突變和擴增[10,28,42]。最近研發的特異性CDK12抑制劑(如THZ531)不僅是強有力的CDK12研究工具,而且更具有腫瘤靶向藥物治療的前景[11]。目前,已經證明THZ531單藥治療對于CDK12過表達和活化的腫瘤(如乳腺癌等)患者是有效的[10]。此外,CDK12特異性抑制劑的研究也可為PARP1/2抑制劑耐藥的腫瘤(如三陰乳腺癌等)進行靶向藥物治療提供一種有效的治療方案。然而,目前關于細胞中CDK12失活或缺失后將引起怎樣的細胞生理、病理反應;某些實體腫瘤(如胰腺癌、胃癌等)是否存在CDK12的異常突變;對化療或靶向治療藥物不敏感、易耐藥的腫瘤組織和細胞中是否廣泛存在CDK12抑制劑耐藥性;改變這些腫瘤組織和細胞中CDK12的表達水平是否能對腫瘤的靶向治療進行分子水平的預測等研究難題,國內、外文獻尚無相關的報道,期待進一步的研究闡明。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
