超聲活化過硫酸鹽氧化降解水溶液中的全氟辛烷磺酸鹽
2019-04-22 06:41:04李炳智
農業環境科學學報 2019年4期
關鍵詞:體系
李炳智,朱 江
(上海市環境科學研究院,上海 200233)
全氟辛烷磺酸及其鹽類(PFOS)是一類新型人工合成的持久性有機污染物(POPs),以其穩定的化學結構及疏水疏油的特點而被廣泛應用于輕水泡沫滅火劑、電鍍鉻霧抑制劑、農藥生產以及油田回采處理劑等生產領域[1]。從20世紀50年代以來,由于PFOS產品的大量生產、使用和生物富集作用,PFOS已呈現出全球分布的態勢。研究證實,在一些環境介質,如地表水[2-3]、土壤[2]、大氣[3]、沉積物[3-4]、地下水[2]、污水處理廠[5],甚至魚類[6]、人體血液[3]和頭發[7]內均檢測出PFOS的存在,它可通過食物和飲用水攝入的方式進入到人體中[2]。鑒于PFOS分布廣、在生態系統中毒性累積性強、難于在環境中降解等獨特性,因此已成為國際上水處理領域繼內分泌干擾物、持久性有機污染物后又一研究熱點。
我國是目前少數生產PFOS類物質的國家之一,也是全球最大的生產國和使用國。我國PFOS生產和使用主要集中在工業發達、新興產業密集的東部沿海地區[8]。據統計,2010年我國PFOS產量仍有100余t,使用量約80萬t[9],PFOS工業源的排放量約為70 t·a-1(占總量70%),排放密度呈現由東向西遞減的趨勢,經濟和工業發展程度最高的京津滬、江蘇、廣東、浙江和山東是PFOS工業源以及生活源排放密度最高的區域[10]。目前我國水環境監測中普遍檢出PFOS,其質量濃度范圍介于未檢出~458 ng·L-1,局部地區PFOS污染水平超過國外[9,11];而且,國內外尚未制訂水環境中PFOS的質量基準/標準。因此,開展我國水環境PFOS的有效降解及其機理研究對于應對全球性PFOS污染,確保我國水環境安全具有重要意義。
PFOS結構中氟具有高的電負性(-4.0),碳原子與之結合形成了穩定的C-F鍵,鍵能可達484 kJ·mol-1[12],常規的生物、化學處理不能將其有效去除和礦化[13-14]。近年來,利用高級氧化和還原技術降解PFOS的研究論文數量呈現穩步上升的趨勢,這說明學術界對水環境PFOS污染防治領域給予高度關注,且取得了良好的降解效果[15-16]。這類技術以不同的方式產生強氧化性、非選擇性的自由基(包括·OH、O-2··和·等),以及高活性、非選擇性還原物質或自由基(水合電子、H·和SO-3·等)為特征,產生的自由基隨即進攻PFOS,使其得到快速分解。針對污染場地地下水環境中普遍存在的1,1,1-三氯乙烷和共存的溶劑穩定劑1,4-二 口惡烷,筆者所在的課題組成功通過優化超聲活化過硫酸鹽工藝參數,使二者達到快速同時去除[17-18]。
鑒于此,本文以新型PFOS為模式污染物,利用構建的超聲活化過硫酸鹽氧化體系,嘗試開展該體系降解水中PFOS的降解影響因素及降解產物研究,考察不同反應體系、超聲頻率、過硫酸鹽投量、初始pH值和空化氣體種類等對PFOS降解速率的影響,并對PFOS降解過程中產生的自由基及其降解中間產物進行捕捉和鑒定,以期為PFOS污染水環境的治理和修復提供技術支撐。
1 材料和方法
1.1 試驗試劑
如無特殊說明,實驗中所用化學試劑均為分析純,未做進一步純化處理。全氟辛烷磺酸鉀(純度98%,阿拉丁試劑有限公司)、全氟羧酸類中間產物標準品(純度≥98.0%,上海甄準生物科技有限公司)、過硫酸鈉(純度≥98.0%,國藥集團化學試劑有限公司)、5,5-二甲基-1-吡咯-N氧化物(DMPO,97%,阿拉丁試劑有限公司)、乙酸銨(98%)、甲醇(≥99.9%)、硝酸(HNO3)和氫氧化鈉(NaOH)均為分析純試劑,試驗用水均采用蒸餾水。HLB(Oasis)固相萃取柱(200 mg/6 mL)購自美國Suplco公司。
1.2 試驗方法
試驗系統由系列超聲波發生器(20、100、200 kHz和400 kHz,輸出電功率為0~100 W,聲能密度2.67 W·cm-2,中科院東海聲學研究所)、超聲換能器、40 mL吹掃反應瓶和SDC-6型恒溫水槽(江蘇佳美儀器制造有限公司)組成。整個系統溫度控制在(30±0.2)℃,反應在有聚四氟乙烯膜襯墊蓋的玻璃瓶內進行。試驗開始前,先將混合均勻的含有一定體積過硫酸鹽母液和PFOS母液的溶液注入一系列玻璃瓶內,注入空化氣體(氬氣、氮氣、氧氣和空氣)20 min,立即旋緊瓶蓋,置于換能器上。開啟超聲波發生器,反應開始計時,超聲輻射一段時間后,取樣測定水溶液中PFOS濃度及降解中間產物等項目。
1.3 分析方法
PFOS及其降解中間產物分析采用固相萃取前處理-超高效液相-質譜(Agilent 6460,配1260液相系統及工作站)分析。色譜柱ACQUITY UPLC BEH C18column(1.7 μm ×2.1 mm ×50 mm),流動相2 mmol·L-1乙酸銨5%甲醇溶液(A)和甲醇(B)的混合液,采用梯度洗脫模式:0~0.5 min,25%B;5.0 min,85%B;5.1~7.0 min,99%B;7.0~9.0 min,99%B勻速下降至25%B;9.0~12.0 min,25%B。流速 0.4~0.55 mL·min-1,柱溫50℃,進樣量10 μL,電離源為電噴霧電離源負源(ESI),毛細管電壓2.5 kV,離子源溫度120℃,脫氣溶劑溫度350℃,掃描模式MRM,碰撞能量7.0~45.0 eV,錐孔電壓15.0~70.0 V。PFOS直接進樣檢測,PFOS 直接進樣檢測線性范圍為 0.1 μg·L-1~10 mg·L-1,最低檢測限為 0.1 μg·L-1。而降解中間產物的分析采用固相萃取前處理后再進樣。凈化程序如下:分別以5 mL甲醇和超純水活化HLB固相萃取小柱;取100 mL水樣調pH為6,以1.0 mL·min-1流經活化后的小柱;用5 mL 20%甲醇水做淋洗液,氮吹,定容至1 mL。本方法對11種全氟化合物水樣的最低檢測限介于0.011~0.089 pg·mL-1,固相萃取的平均加標回收率范圍為72.6%~144.7%,相對標準偏差介于1.2%~9.2%之間。
以DMPO為自由基誘捕劑,采用德國Bruker EMX-8/2.7型電子順磁波譜儀半定量分析反應體系產生DMPO-自由基加合物信號。測試條件為:共振頻率9.77 GHz,微波功率20.02 mW,調制頻率100 kHz,調制振幅1.0 G,掃探寬度200 G,時間常數40.96 ms,掃描時間83.87 s,接收增益為10 000。
氟離子濃度采用美國戴安公司ICS-3000型離子色譜儀進行測定,采用IonPac AS15陰離子交換分析柱(4.0 mm×250 mm),IonPac AG15保護柱(4.0 mm×50 mm),內置數字化電導檢測器,KOH淋洗液,梯度淋洗模式:0~17.0 min,3 mmol·L-1;17.1~27.0 min,60 mmol·L-1;27.1~31.0 min,3 mmol·L-1,流速 1.2 mL·min-1,進樣體積200 μL。
2 結果與討論
2.1 超聲活化過硫酸鹽體系降解PFOS的效果
預試驗首先考察了超聲活化過硫酸鹽體系(US+PS)降解PFOS的效果,并與空白(Control)、單獨過硫酸鹽氧化(PS)以及單獨超聲處理(US)的效果進行了比較。試驗條件如下:PFOS初始濃度約為10 mg·L-1,過硫酸鹽與PFOS摩爾比為50∶1,初始溶液pH值為7.0,溫度30℃,超聲頻率為400 kHz,功率100 W和超聲密度2.67 W·cm-2,反應時間設為8 h。PFOS降解試驗結果和擬合結果分別如圖1和表1所示。
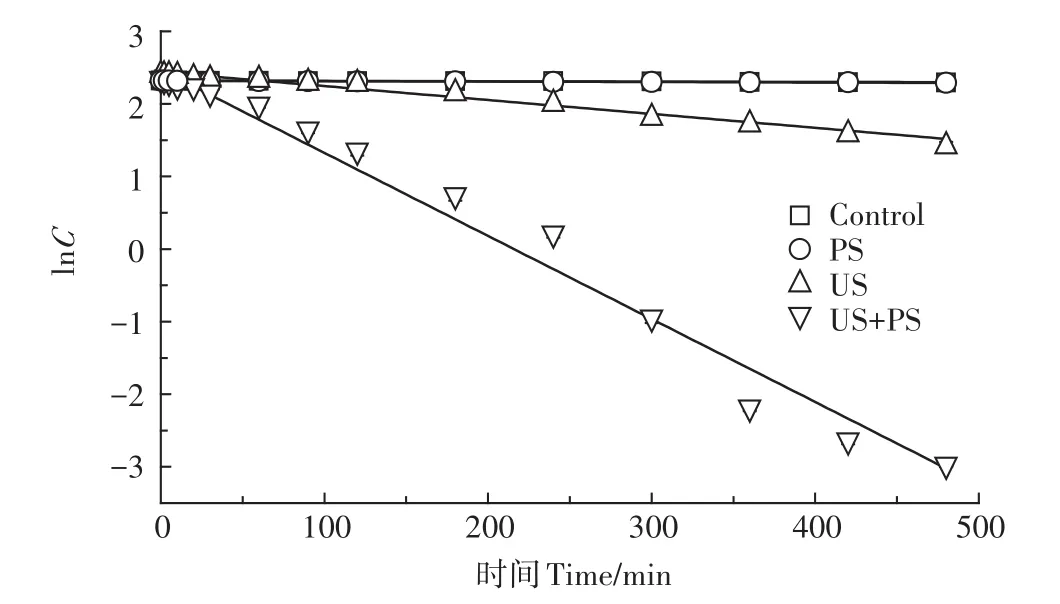
圖1 PFOS在不同反應體系降解的濃度對數-時間曲線Figure 1 Logarithm concentration versus time curve of PFOS degradation at different systems

表1 不同體系中PFOS降解的表觀準一級反應速率常數值Table 1 Pseudo-first order reaction rate constants of PFOS at different systems
由圖1和表1可知,PFOS在4個體系中的降解較符合表觀準一級反應動力學模型:ln(Ct/C0)=-kt,R2≥0.941。在Control和PS體系中PFOS幾乎無任何降解,在整個反應時間范圍內最大降解率低于3%;相比之下,在過硫酸鹽未投加情況下,反應速率亦較低,8 h內PFOS降解率僅為62.7%;然而,當超聲體系投加過硫酸鹽后,8 h內PFOS去除效果顯著改善,達到了最高的降解率(99.5%)。據此推測,超聲體系投加過硫酸鹽后可能生成了更多數量的高活性自由基(SO-4·、·OH、O-2·等),從而極大加速了PFOS的降解和轉化。為了更直觀地評價超聲活化過硫酸鹽體系對PFOS降解的強化效果,這里引入增強因子(R)概念,R定義如式1所示。
R=超聲活化過硫酸鹽體系PFOS降解速率/(單獨過硫酸鹽體系PFOS降解速率+單獨超聲體系PFOS降解速率) (1)
由表1計算得到的超聲活化過硫酸鹽體系降解PFOS的增強因子為5.77,這表明超聲活化過硫酸鹽體系存在顯著的協同效應。
為了證實有活性自由基參與了超聲活化過硫酸鹽體系PFOS的降解,采用DMPO作為自由基的誘捕劑,利用DMPO與自由基加成反應生成較穩定的加合物這一特性,通過電子自旋共振波譜技術可半定量捕捉到加合物信號,克服了自由基壽命短暫難以捕獲的缺點,從而得到直接的試驗證據。利用這一技術,我們獲得了單獨過硫酸鹽體系(PS)、單獨超聲體系(US)和超聲活化過硫酸鹽體系(US+PS)3個體系反應10 min時的DMPO-自由基加合信號,如圖2所示。
由圖2可以清晰地發現,在US體系中得到了DMPO加成物的特征四重分裂峰(實心圓點),峰高比1∶2∶2∶1,其超細常數 αN=14.4 G,αH=15.6 G,g值為2.005 5,這些參數與以前的文獻報道相一致[19]。這是DMPO與羥基自由基加合的特征參數,由此表明US體系生成了羥基自由基。而在PS體系中,還得到了DMPO加成物的特征六重分裂峰(實心三角),其超細常數αN=13.2 G,αH=9.6 G,αH=1.48 G和αH=0.78 G,與文獻報道[20]的硫酸根自由基與DMPO加合物的特征參數相符合,這表明PS體系中存在硫酸根自由基。在“US+PS”體系中,則出現了這兩種自由基的疊加峰,而且從加合物的信號強度來看,顯著高于PS和US體系之和,這一現象直接解釋了“US+PS”體系存在顯著協同效應的原因:正是這些明顯增多的硫酸根和羥基自由基導致了PFOS的快速降解。
2.2 超聲頻率對PFOS降解的影響
已有研究證實PFOS在活化過硫酸鹽和高頻超聲作用下能夠降解主要歸因于活化過硫酸鹽產生的高活性物質以及高頻超聲誘發的較強化學效應,且其降解均遵從表觀準一級反應動力學模型[21-23]。本文選擇20、100、200 kHz和400 kHz 4個超聲頻率進行試驗,其他試驗條件均為:初始PFOS濃度約為10 mg·L-1,過硫酸鹽與PFOS摩爾比為50∶1,初始pH值為7.0,溫度30℃。表2給出了4個超聲頻率下PFOS降解的表觀準一級反應動力學速率常數。由表2可知,PFOS在4個不同超聲頻率的降解行為均遵從表觀準一級反應動力學規律。隨著超聲頻率的升高,PFOS降解的表觀準一級反應速率逐漸增大,由50 kHz時的8.313×10-4min-1增至400 kHz時的1.144×10-2min-1。
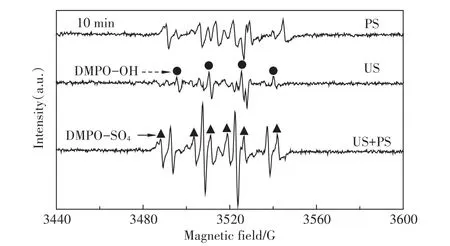
圖2 10min時PS、US和US+PS體系DMPO-自由基加合ESR信號Figure 2 ESR spectra of PS,US,and US+PS in the presence of 0.09 mol·L-1DMPO after reaction for 10.0 min
超聲頻率的影響與空化泡的形成動力學有關,較高超聲頻率能夠實際增加反應體系自由基的數量。較高超聲頻率時空化泡破滅過程雖不如低頻時那樣劇烈,但空化過程周期縮短,單位時間內產生的超聲空化效應更多,從而更有可能生成更多數量的自由基[24]。由于較高頻率時空化泡壽命縮短,自由基在發生自終止反應前有更大幾率從空化泡逃逸并遷移至液相本體[25]。
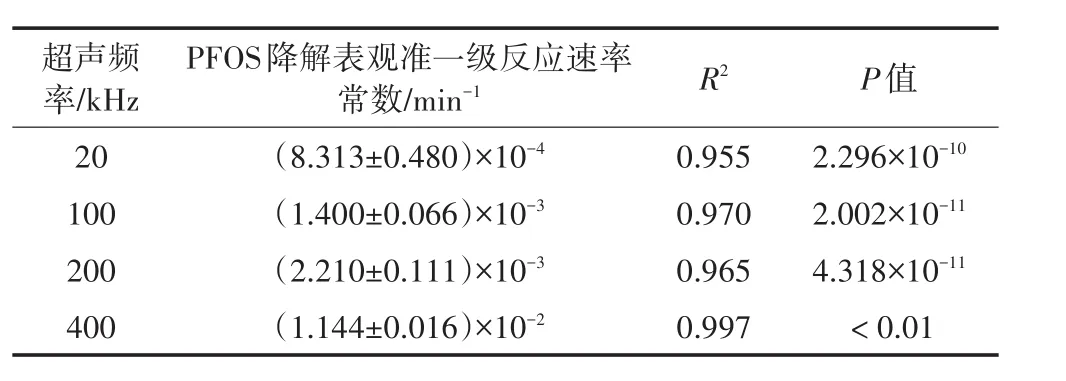
表2 不同超聲頻率下PFOS降解的表觀準一級反應速率常數值Table 2 Pseudo-first order reaction rate constants of PFOS at different ultrasonic frequency
2.3 過硫酸鹽濃度對PFOS降解的影響
控制PFOS初始濃度約為10 mg·L-1,初始pH為7.0,溫度30℃,超聲頻率為400 kHz,功率100 W和超聲密度2.67 W·cm-2,過硫酸鹽投加濃度按與PFOS初始濃度的摩爾比1∶1、5∶1、20∶1、50∶1和100∶1投加,試驗研究了過硫酸鹽投加濃度對PFOS降解性能的影響。PFOS濃度對數值與時間的擬合試驗結果如表3所示。
如表3所示,不同過硫酸鹽濃度時PFOS的降解均符合表觀準一級反應動力學模型,且與實驗數據擬合良好(R2>0.969)。當過硫酸鹽與PFOS摩爾比由1∶1增加至50∶1時,PFOS降解的表觀準一級反應速率常數由2.470×10-3min-1增至1.144×10-2min-1。然而,當二者摩爾比進一步增大至100∶1時,表觀準一級反應速率常數不升反降。
為了探求不同過硫酸鹽投量下PFOS降解的表觀準一級反應速率常數下降的原因,試驗利用ESR測定了不同過硫酸鹽投量時DMPO與自由基加合物信號強度的變化,試驗結果如圖3所示。
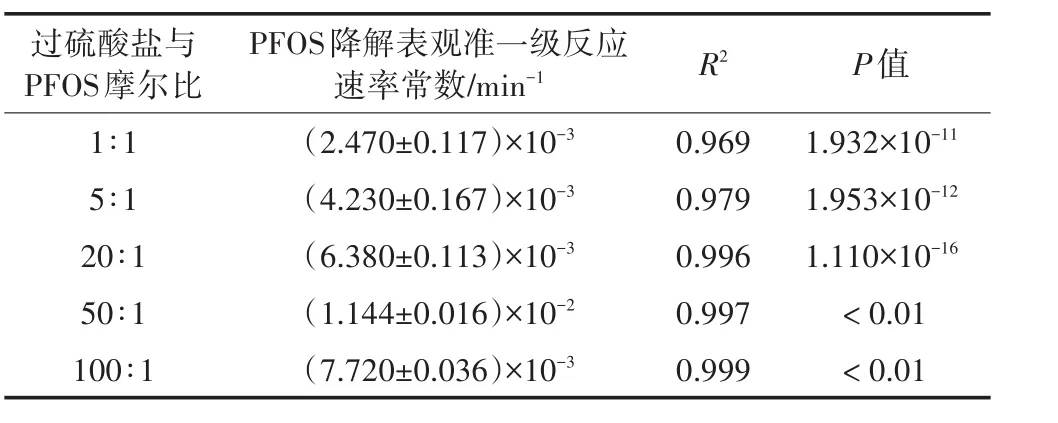
表3 不同過硫酸鹽濃度時PFOS降解的表觀準一級反應速率常數值Table 3 Pseudo-first order reaction rate constants of PFOS at different persulfate dosages
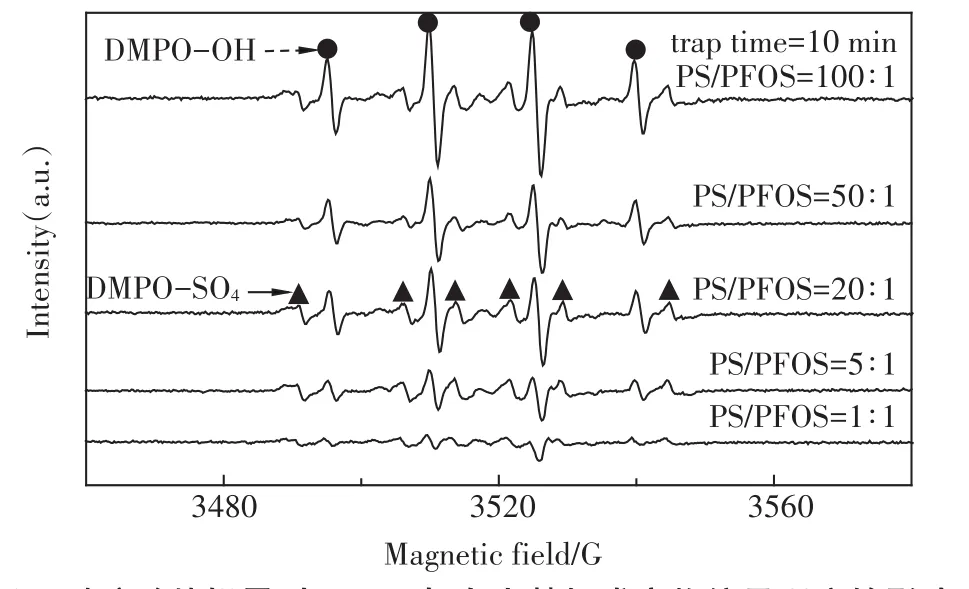
圖3 過硫酸鹽投量對DMPO與自由基加成產物信號強度的影響Figure 3 Effect of persulfate dosage upon the intensity of the formation of DMPO adducts
由圖3可以看出,過硫酸鹽與PFOS摩爾比從1∶1增加至100∶1時,DMPO-SO4以及DMPO-OH峰強度和自由基濃度也相應增加,但試驗結果中卻未出現與此相匹配的PFOS強化降解現象。據此推測,可能的原因有:
(1)硫酸根自由基和羥基自由基一旦生成即隨之發生一系列化學反應,包括活性物質的形成、自由基重組以及過硫酸鹽猝滅反應,如方程式2~方程式10所示[26]。盡管羥基自由基(E0=+2.7 V)有比硫酸根自由基(E0=+2.6 V)較高的氧化還原電位值[27],但伴隨硫酸根自由基生成而產生的二價硫酸根離子卻會抑制上述兩種自由基的反應活性[28]。

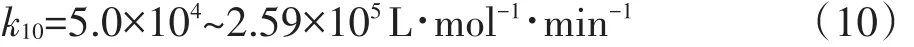
(2)硫酸根自由基自身的重組反應(式5)、羥基自由基自身重組反應(式4)以及二者之間的反應(式8)速率比羥基自由基、硫酸根分別與PFOS的反應以及過硫酸鹽的猝滅反應(式9、式10、式6和式7)約高1~3個數量級[16,29-31]。由于自由基之間的競爭性消耗作用,當過硫酸鹽投加過量時這些反應會導致自由基的利用率低下。(3)其他也有類似研究表明,對于有機物氧化而言,的確存在一個最佳的過硫酸鹽投加值,如高于此臨界值,污染物的降解就會受到一定程度的抑制[32-34]。
2.4 反應體系初始pH值對PFOS降解的影響
為了考察初始pH值變化對超聲活化過硫酸鹽體系降解PFOS速率的影響,控制試驗條件為:初始PFOS濃度約10 mg·L-1,初始pH值分別為3.0、7.04和11.03,溫度30℃,超聲頻率400 kHz,功率100 W、超聲密度2.67 W·cm-2,過硫酸鹽與PFOS初始濃度的摩爾比為50∶1。PFOS濃度對數值與時間的表觀準一級反應動力學擬合試驗結果如表4所示。
如表4所示,隨著溶液初始pH值由3.0增至11.03,相應的PFOS降解的表觀準一級反應速率常數由0.013 75 min-1下降至0.009 47 min-1。之所以出現上述試驗現象,這可能是因為:(1)酸性條件時,由于酸的催化作用會生成額外多的硫酸根自由基[28];(2)在堿性條件時,硫酸根自由基會與氫氧根離子反應生成更多的羥基自由基,如方程式(3)所示;羥基自由基通常通過H原子抽提進攻有機物分子產生水,但PFOS分子本身不含H原子,無法發生H原子抽提反應,水溶液中二者間的反應活性較差,從而減慢了PFOS的降解速率,故堿性條件不利于硫酸根自由基對PFOS的降解。隨著反應的進行,盡管溶液pH值逐漸下降(例如初始pH值分別為3.00、7.00和11.03時,經過近500 min,反應終了pH值逐漸分別降至2.70、6.42和10.38),但是溶液初始pH值越高,氫氧根離子濃度越高,被其消耗的硫酸根自由基也越多,從而削弱了反應體系的氧化作用。
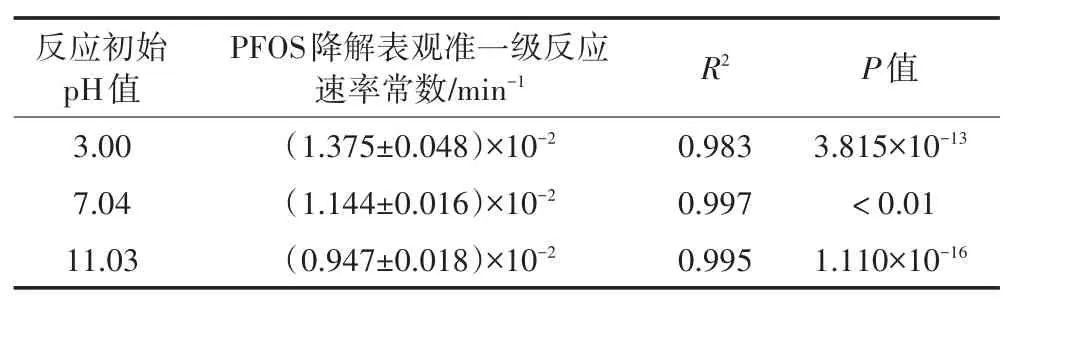
表4 不同初始pH值時PFOS降解的表觀準一級反應速率常數值Table 4 Pseudo-first order reaction rate constants of PFOS at different initial pH values
2.5 空化氣體種類對PFOS降解的影響
空化氣體種類(如氮氣、氧氣、空氣和氬氣等)對超聲的影響主要表現為反應體系活性物質(如H·、·OH、H2O2、H2、HO2·等)種類及其產率的差異,進而對污染物的降解行為產生影響[35]。4種空化氣體對超聲活化過硫酸鹽體系降解PFOS速率的影響結果,見表5。
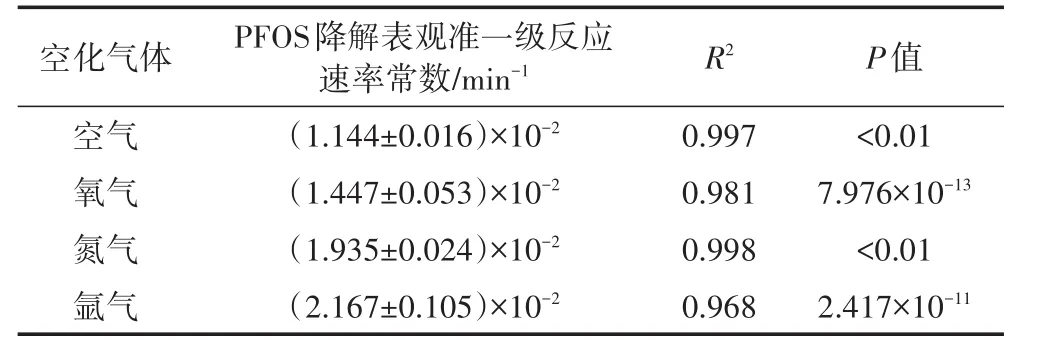
表5 不同空化氣體PFOS降解擬合的表觀準一級反應速率常數值Table 5 Pseudo-first order reaction rate constants of PFOS at different cavitation gas
由表5可知,4種空化氣體(氮氣、氧氣、空氣和氬氣)存在下,PFOS降解均遵從表觀準一級反應動力學,反應速率常數大小順序為:空氣<氧氣<氮氣<氬氣。之所以出現上述結果,其原因可能是:(1)比熱大的空化氣體更有利于空化氣泡的崩潰,由空化效應獲得的聲化學效應越大,單原子氣體比雙原子氣體、雜原子氣體更適合作為空化過程的氣源[36];(2)空化氣體導熱率越大,空化氣泡崩潰過程積累熱量將更多地傳給周圍液體,從而降低最高溫度值;(3)溶解度大的空化氣體會降低空化閾值和空化強度[37]。根據上述試驗結果可知,PFOS降解效果最佳的空化氣體為氬氣。
目前,雖然高級氧化(臭氧氧化、電化學氧化、超聲、過硫酸鹽氧化和光解)和還原工藝(零價鐵和檸檬酸鈦)處理水中PFOS的研究報道較多,但由于這些研究中PFOS初始濃度范圍設置較為寬泛,使得很難客觀地比較這些方法在降解PFOS性能上的優劣[16]。文獻報道中單獨超聲(200 kHz,200 W)和50℃熱活化過硫酸鹽分別降解 20 μmol·L-1的PFOS,其一級反應速率常數分別為0.016 min-1和0.000 47 min-1[16],與本文中PFOS降解速率常數基本相當。高效超聲換能器研發成功之前,綜合過硫酸鹽和超聲兩種AOPs工藝的耦合協同作用提高對PFOS的降解效率,起到“1+1>2”增效作用的同時顯著降低了超聲能耗。
2.6 降解中間產物鑒定及可能的降解機理
為了深入研究PFOS在超聲活化過硫酸鹽體系中的降解機理,試驗采集了不同反應時間間隔內的PFOS溶液樣品,并將其混合。利用超高效液相色譜-質譜儀(UPLC-MS)捕捉到了一系列PFOS降解產物,其典型色譜圖如圖4所示。
根據與全氟代標準物質色譜圖比對,確認檢測到的8種物質及停留時間(Rt)分別為:全氟乙酸(Rt=1.22 min)、全氟丙酸(Rt=1.27 min)、全氟丁酸(Rt=2.49 min)、全氟戊酸(Rt=3.13 min)、全氟己酸(Rt=3.73 min)、全氟庚酸(Rt=3.79 min)、全氟辛酸(Rt=4.18 min)、未知物質(Rt=4.45 min)和PFOS(Rt=4.55 min)。
有關超聲和活化過硫酸技術降解PFOS產物研究數量相當有限。日本大阪府立大學Moriwaki等[38]利用200 kHz超聲降解PFOS的產物包括:三氟乙酸、五氟丙酸、七氟丁酸、九氟戊酸、全氟己酸、全氟庚酸、全氟己烷磺酸和全氟辛酸等;楊佘維等[39]發現,真空紫外耦合高頻超聲體系中PFOS降解中間產物為7種短鏈全氟羧酸(三氟乙酸、五氟丙酸、七氟丁酸、九氟戊酸、全氟己酸、全氟庚酸和全氟辛酸)以及3種短鏈全氟磺酸(全氟庚烷磺酸、全氟己烷磺酸和全氟丁烷磺酸)。由此可見,文獻報道的PFOS降解產物與本文研究結果基本一致,即PFOS在反應體系中主要分解為兩類物質:短鏈的全氟代羧酸類和全氟代磺酸類中間產物。
超聲活化過硫酸鹽體系降解有機化合物主要遵從熱解和(或)自由基(如SO-4·、·OH等)誘導的反應機制。PFOS較高的辛醇-水分配系數、亨利系數和蒸汽壓使其具有較好的親水性和較低的揮發性,因此可以預知:反應初始階段其可能在空化泡內或空化泡-溶液交界面處進行熱解反應,以及在液相本體中進行自由基的氧化反應[40]。結合分析測定的中間產物,超聲活化過硫酸鹽體系降解PFOS可能的機理見圖5。標框的物質為本研究檢測到的中間產物。
文獻報道PFOS鉀鹽的pKa值為-3.5[41],在所有可能的環境條件(強酸性條件除外)下,PFOS鉀鹽均以離子狀態存在。根據檢測到的中間產物和相關文獻報道,推測可能的反應機理有以下兩種途徑:(1)水相本體反應。在超聲場和硫酸根自由基的誘導下,PFOS陰離子首先被提取一個電子,形成C8F17SO3·自由基,然后脫去磺酸基,生成C8F17·自由基。C8F17·自由基末端的碳原子具有較高的電荷密度,極易與氧化劑反應生成熱不穩定全氟代醇類物質C8F17OH;C8F17OH脫去氟化氫而生成酰基氟C7F15COF,C7F15COF極易水解,生成少1個CF2單元的C7F15COOH。然后SO-4·自由基進攻C7F15COO-陰離子,轉化為硫酸鹽、二氧化碳和C7F15·;C7F15·重復上述成醇、脫氟化氫和水解步驟,短鏈的C2~C7全氟羧酸降解產物會依次產生,最終PFOS逐步降解成氟離子和二氧化碳。(2)氣泡-水界面反應。首先PFOS被吸附到氣泡-水的界面,通過超聲產生局部的高溫高壓效應,PFOS末端的C-S鍵熱解斷裂,釋放氣體SO3至水相,自身轉化為氣態全氟烯烴;氣態全氟烯烴在高溫條件下,裂解為氟化物自由基,在自由基和高溫的進一步作用下繼續轉化為二氧化碳、一氧化碳和氟化氫。
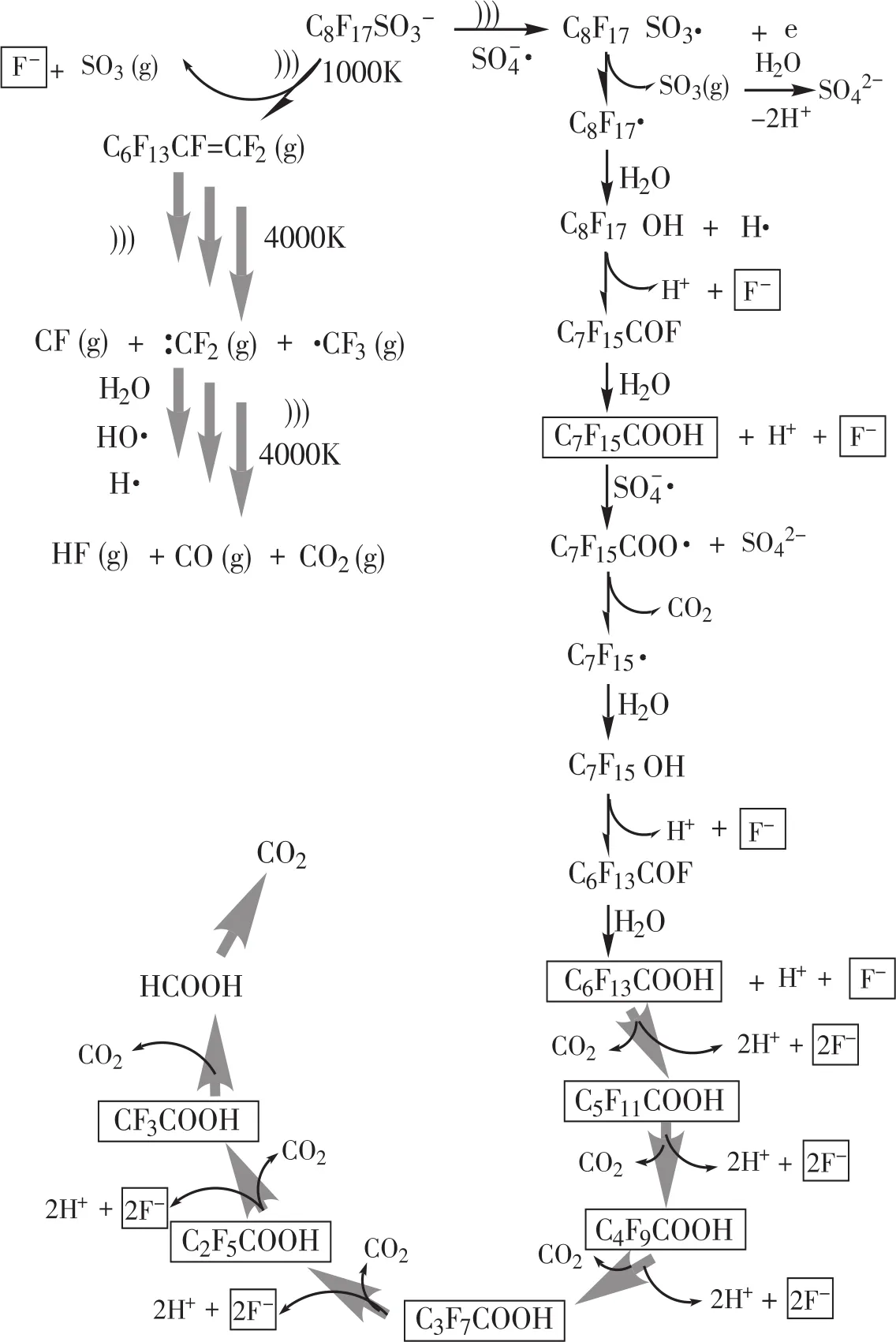
圖5 PFOS在超聲活化過硫酸鹽氧化體系中可能的降解途徑Figure 5 Proposed PFOS degradation pathway in the sono-activated persulfate oxidation process
由PFOS的降解途徑可知,PFOS的降解主要在氣相反應和液相反應作用下逐漸分解為短鏈的全氟羧酸和全氟磺酸中間產物;然而,有相當多的研究表明,這些中間產物的降解速率要慢于母體污染物PFOS[16]。例如,在本文的試驗條件下,PFOS經過8 h基本完全降解,而其脫氟率僅為90%,這說明仍有部分含氟PFOS降解中間產物尚未得到完全脫氟。在PFOS污染水體處理實踐中,PFOS母體污染物的完全降解并不是處理的終點;在此之后仍需繼續延長處理時間,以使得PFOS降解中間產物(如短鏈全氟羧酸和全氟磺酸類等)得到徹底降解。
關于世界上其他國家和地區水環境中PFOS的濃度值,已有文獻報道。PFOS在地表水、污水處理廠出水和飲用水中濃度分別介于1~7、7~50 ng·L-1和1~100 ng·L-1[42],基本與我國水環境中PFOS的檢測濃度相當。為此,一些國家已制定飲用水中全氟化合物健康指導值為0.04~0.5 μg·L-1[43]。本文之所以設置的PFOS模擬濃度較環境介質報道值高,是為了便于檢測濃度較低的PFOS降解產物,以更好地闡釋其降解機理。后續研究將基于我國環境介質中PFOS檢出濃度開展有針對性的PFOS氧化和還原降解的對照研究。
3 結論
(1)與單獨超聲和單獨過硫酸鹽氧化體系相比,超聲活化過硫酸鹽體系能夠顯著加速PFOS的降解和轉化,存在明顯的協同效應,增強因子達到5.77,其本質原因在于:協同體系生成了明顯增多的·OH和SO-4·。
(2)該體系降解PFOS(初始濃度約10 mg·L-1)較佳的實驗條件為:溫度30℃、超聲頻率400 kHz、功率100 W、超聲密度2.67 W·cm-2、過硫酸鹽與PFOS摩爾比為50∶1,初始pH值為7.0,空化氣體為氬氣,反應時間為8 h。
(3)根據檢測到的7種PFOS降解產物,給出了PFOS在超聲活化過硫酸鹽體系中可能的降解途徑。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
