基于XGBoost的RNA修飾位點的識別
2019-05-25 11:26:18呂成偉樊永顯
桂林電子科技大學學報 2019年6期
關鍵詞:模型
呂成偉, 樊永顯
(桂林電子科技大學 計算機與信息安全學院,廣西 桂林 541004)
自經過修飾的RNA核糖核酸第一次發現以來,被人類已知的RNA修飾類型已經達到了約150種[1]。研究表明,RNA修飾是基因調控的關鍵組成部分[2],其參與了轉錄后的各種生物過程,如蛋白質翻譯和定位、mRNA剪接等,并發揮著重要作用[3]。但是,RNA修飾在其他方面的功能對人們來說仍然是未知的。因此,預測RNA修飾位點對于理解它們的分子機制和功能起著至關重要的作用。
新一代測序技術的出現為在全基因組范圍內研究RNA修飾提供了契機。如N1-甲基甘氨酸(m1A)、N6-甲基甘氨酸(m6A)和5-甲基胞嘧啶(m5C)圖譜可用于人類轉錄組。雖然這些基于生物實驗技術的高通量測序方法在理解生物功能和RNA修飾方面起到了積極推動的作用,但這些方法有很大局限性,其實驗成本高、耗時長。為了解決該問題,一些用于識別RNA修飾位點的基于高分辨率實驗數據的計算方法被提出。針對m6A修飾位點的識別問題,Chen等[4]提出了基于序列的iRNAMethy方法,使用了偽二核苷酸組分(pseudo dinucleotide composition,簡稱PseDNC)編碼方式,在特征提取方式上取得了一些突破。Chen等[5]在編碼方式上進行了創新,提出了m6Apred方法,在原有序列信息的基礎上計算出核苷酸的頻率信息,且加入了其化學分類特征,進一步提高了m6A修飾位點的識別準確率。Zhang等[6]提出了一種新的提取特征的方法m6A-HPCS,該方法的思想與Chen等[5]提出的m6Apred基本一致,從23種核苷酸物理化學性質中出尋找一個最優子集,結合自協方差和互協方差變換提取序列特征。
上述幾種方法為基于序列的RNA修飾位點的預測開辟了道路,并取得了一定的成功。但這幾種方法對m6A和m5C修飾位點的識別準確率不夠理想,仍有較大的提升空間。針對上述問題,在采用PseKNC的編碼方式對樣本序列進行編碼的基礎上,添加了位置特異性單核苷酸及二核苷酸偏好特征,并基于XGBoost集成算法構建了預測模型。實驗證明,該模型的魯棒性好,且針對上述提出的3種RNA修飾位點的預測都取得了較高的準確率。
1 材料與方法
為了開發一種新的預測方法,文獻[7-11]遵循了Chou[12]提出的一些原則,并明確以下4個步驟:
1)構造一個高質量的基準數據集用來訓練和測試預測模型;
2)對生物序列進行編碼,使其能夠被預測模型識別;
3)選擇或者自主研發一種魯棒的算法來建立預測模型;
4)進行交叉驗證,并客觀地評價預測模型的好壞。
1.1 基準數據集
本研究所使用的數據集包括m1A、m6A、m5C 3種RNA序列[13],這3種RNA序列分別包含6 366、1 130和120個正樣本。為了平衡正負樣本,從對應的負樣本中隨機選取了6 366、1 130和120個樣本分別作為m1A、m6A和m5C的負樣本。
1.2 構建RNA序列樣本
生物信息學面臨的一個極具挑戰的問題是:如何對生物序列進行有效編碼,使得其編碼后的序列盡可能包含序列模式特征,并能被現有的機器學習模型直接識別。幾乎所有的機器學習算法都是為了處理向量而開發的,并不能直接識別序列樣本。常用的獨熱編碼雖然能解決不能被機器學習模型識別的問題,但這種編碼方式會丟失許多重要的序列模式特征,從而導致最終的預測模型預測能力低下。為了解決蛋白質序列的問題,研究人員開發了偽氨基酸組成物(pseudo amino acid composition,簡稱PseAAC)[14-18]。自PseAAC引入以來,它的概念幾乎滲透到計算蛋白質組學的每個領域。在PseAAC概念的啟發和巨大成功的鼓舞下,PseKNC[28]被提出來,并應用到基因組分析的各個領域。
假設RNA序列由L個核苷酸殘基組成,
S={R1,R2,…,RL},
(1)
其中R1、R2分別為RNA序列的第1、2個核苷酸殘基。
本研究用PseKNC對RNA序列進行編碼,最終得到具有4k個分量的向量,即
(2)
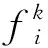
對于式(2),當k=2時,表示RNA序列由二核苷酸配置而成,于是有
D=[f(AA)f(AC)f(AG)…f(UU)]T=
(3)
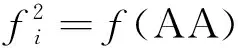
D=[f(AAA)f(AAC)f(AAG)f(UUU)]Τ=
(4)
1.3 XGBoost
XGBoost是一種基于GBDT(gradient boosting decision tree,簡稱GBDT)梯度下降框架的集成學習算法。GBDT是將梯度下降和決策樹相結合,基于前一個分類器殘差減少的方向上,構造新的分類器,通過多次迭代構造一組弱分類器,對弱分類器輸出結果進行加權累加,累加結果作為強分類器輸出[21]。XGBoost與GBDT相比,其優點在于改變了GBDT基于Boosting串行序列化求解問題的方式,利用CPU多線程分布式并行計算,并通過對殘差進行泰勒二次展開進行求解,從而打破了現有庫的計算速度和精度,使得數據處理和運算的速度得到了提升。
造成XGBoost模型精度高、運行速度快的另一個因素是選用分類回歸樹(classification and regression tree,簡稱CART)作為決策樹。由于CART樹的葉子節點對應的值是一個實際的分數,而非一個確定的類別,這使得優化算法的實現變得更加高效。將XGBoost模型表示為如下數學形式:
(5)
其中:F為所有可能的CART樹的集合;f為一棵具體的CART樹;K為樹的棵數。該XGBoost模型由k棵CART樹組成。
1.4 性能評估
通過如下步驟判斷一個模型的優劣:
1)采取交叉驗證的方法測試模型。如文獻[22]采用交叉驗證的方法對模型進行測試,得到了廣泛的認可和使用。交叉驗證對于本實驗的模型測試也同樣不失為一種好的方法。為了減少隨意性,并使得實驗結果更準確,采用10次十折交叉驗證。
2)采用Chou[12]在研究信號肽預測中使用的4個度量參數評價模型。根據文獻[8,23-24]中的定義,敏感性N、特異性P、準確率A和馬修斯相關系數M分別表示為:
(6)
其中:NTP為含有修飾位點的樣本被正確預測為含有該修飾位點數;NTN為不含有修飾位點的樣本被正確預測為不含有該修飾位點數;NFP為不含有修飾位點的樣本被錯誤預測為含有該修飾位點數;NFN為含有修飾位點的樣本被錯誤預測為不含有該修飾位點數。
1.5 位置特異性核苷酸偏好特征
位置特異性偏好思想在生物信息學得到了廣泛應用,在功能位點的識別及啟動子位點的識別方面都取得了非常不錯的效果[25-27]。其原理是統計生物序列中某些關鍵位置或某種核苷酸出現的概率[26],將得到的概率值作為位點識別的特征。受此啟發,將位置特異性單核苷酸和雙核苷酸偏好特征特征應用到RNA修飾位點中。
1.5.1 位置特異性單核苷酸偏好特征
由式(1)可知,每個樣本由L個核苷酸組成,對于一個基準數據集的所有樣本,可分別計算出第j(j=1,2,…,L)個位置上4種核苷酸出現的概率,并用一個長度為4的向量表示:
(7)
其中,MA,j、MC,j、MG,j和MU,j分別為A、C、G和U這4種核苷酸在第j個位置出現的概率。
將j從1取值到L得到的位置特異性向量Mj組合在一起,構成一個4×L維的位置特異性單核苷酸偏好矩陣M:
(8)
1.5.2 位置特異性雙核苷酸偏好特征
取2個相鄰的核苷酸為一個單元,則式(1)樣本序列可表示為
S′={N1,N2,…,NL-1},
(9)
其中,Nj={Rj,Rj+1},j=1,2,…,L-1表示第j個位置雙核苷酸的類型,而雙核苷酸的種類共有16種,即Nj∈{AA,AC,AG,AU,CA,…,UU}。
與M的計算過程類似,可計算出一個維度為16×(L-1)的位置特異性雙核苷酸偏好矩陣:
(10)
1.6 特征選擇
雖然用PseKNC對RNA序列進行編碼是一個不錯的選擇,但這種編碼方式有一個缺點,即數據樣本維度會出現爆炸式的增長,從而會出現以下問題:1)容易造成過擬合導致模型的泛化能力偏低;2)信息冗余和噪聲會導致模型準確率低下,達不到預期效果;3)高維度樣本使得計算機的運算負荷加重,從而使模型的運行時間大大增加。
為了解決上述問題,對數據樣本進行特征選擇,進而降低樣本維度。本研究采用F-score[28]特征選擇方法,定義如下:
(11)
2 結果
2.1 PseKNC編碼方式中最優k值的確定
對m1A、m6A、m5C這3種RNA序列采用PseKNC的編碼方式進行編碼,但k取不同的值時,結果也不同,為了找到最優的k值,對k=2,3,4,5,6,7時分別進行編碼,其十折交叉驗證的結果如圖1所示。
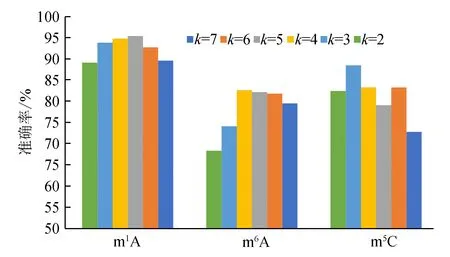
圖1 PseKNC編碼中不同的k值對m1A, m6A 和 m5C修飾位點的識別準確率的影響
從圖1可看出,k取不同值時,m1A、m6A、m5C這3種RNA修飾位點的識別準確率也隨之波動,其中m1A對于不同的k值波動較小,而k值的不同對m6A和m5C的影響較大。當k=5時,m1A修飾位點的識別準確率最高;k=4時,m6A修飾位點的識別準確率最高;k=3時m5C修飾位點的識別準確率最高。在進行特征選擇之前,m1A、m6A、m5C的PseKNC編碼中k的最優值分別為5、4、3。
為了進一步提高識別準確率,采用F-score特征選擇方法得到k最終的最優解。
在m1A、m6A、m5C取不同k值(k=2,3,4,5,6,7)編碼后的序列基礎上,融合位置特異性單核苷酸偏好特征和位置特異性雙核苷酸偏好特征,再對其進行特征選擇。十折交叉驗證的結果如表1所示。
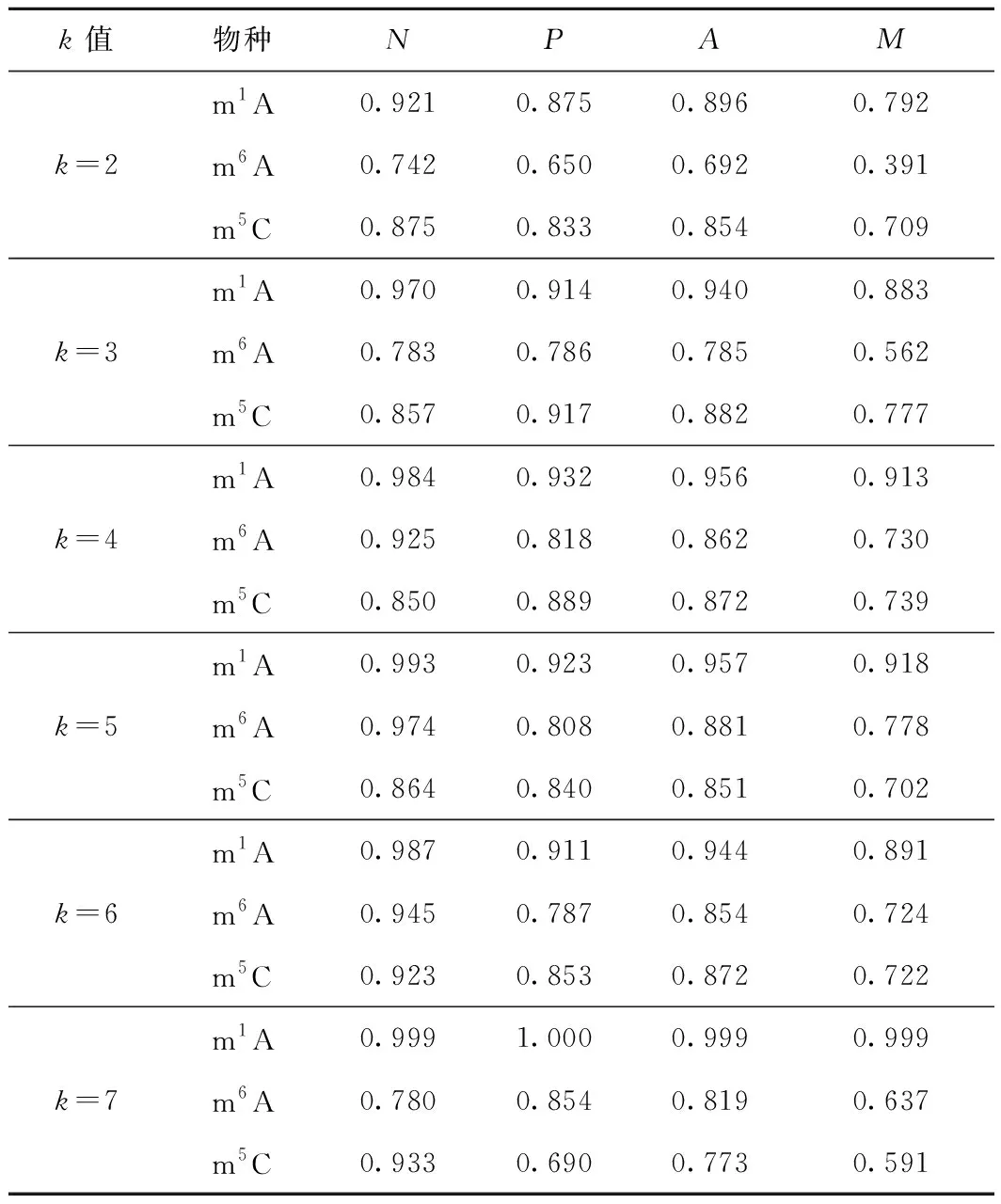
表1 特征選擇后,不同的k值對m1A、m6A、m5C修飾位點的識別結果
從表1可看出,融合了位置特異性單核苷酸偏好特征和位置特異性雙核苷酸偏好特征并經過特征選擇后,m1A、m6A、m5C這3種RNA修飾位點的識別準確率均有較大提升。其中:m1A在k=7時,識別準確率達到最高,為99.9%;m6A在k=5時,識別準確率達到最高,為88.1%;m5C在k=3時,識別準確率達到最高,為88.2%。而在此之前,m1A、m6A、m5C這3種RNA修飾位點的識別準確率最高時其PseKNC編碼對應的k值分別為5、4、3,顯然,除了m5C的PseKNC編碼中最優k值未發生改變,其他2個均發生了改變。
2.2 基于網格搜索的XGBoost模型參數尋優
在確定了PseKNC編碼的最優k值后,繼續對XGBoost預測模型的參數進行尋優。要想完全發揮XGBoost的強大性能,對其進行調參是必不可少的一項工作。這里選用網格搜索的方法對其進行調參。網格搜索的原理是:在所有候選的參數中,通過循環遍歷,嘗試每種可能性,交叉驗證后,表現最好的參數組合就是最終結果。該方法的優點是結果準確,但缺點是當參數數量過多時,參數尋優的計算過程非常耗時。XGBoost中的參數主要分為通用參數、學習任務參數和命令行參數3大類。其中通用參數有20多個,學習任務參數有4個,命令行參數有十多個,若對這些參數進行網格搜索尋優,將會非常耗時,使調參的工作面臨巨大挑戰。
為了應對這一挑戰,針對性地選取一些核心參數,將對模型性能影響不大的參數剔除。據此,在通用參數中選取booster、learning_rate、max_depth、min_child_weight、subsample、colsample_bytree、gamma、lambda和alpha這8個參數,在學習任務參數中選取objective參數,在命令行參數中選取num_round參數。其中:max_depth和min_child_weight對決策樹的構建起約束作用;subsample和colsample_bytree均是關于采樣的參數;lambda和alpha均是正則項。為了進一步減少運算量,采用組合分批網格搜索的策略,即將具有相同作用的參數組合在一起,對組合進行網格搜索,將最優組合與其他參數組合在一起,再進行網格搜索。調參后的XGBoost模型在jackknife測試下的識別結果如表2所示。從表2可看出,m6A修飾位點的識別準確率從88.1%提升到了92.6%,m5C修飾位點的識別準確率從88.2%提升到了89.6%,雖然m1A修飾位點的識別準確率并未得到提升,但在此之前已經達到了99.9%這樣一個非常理想的水平。
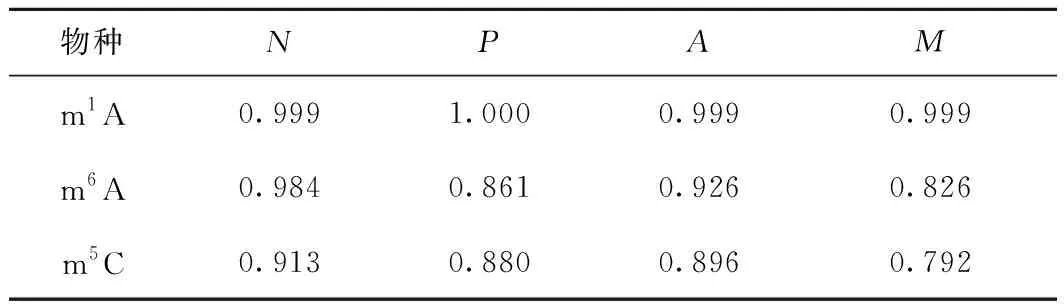
表2 參數調整后的XGBoost模型的識別結果
ROC(receiver operating characteristic)曲線是一個能直觀展現模型性能的另一個重要指標[29]。ROC曲線下側包含的面積(AUC)越大,模型的性能越好[30]。圖2為XGBoost預測模型分別對m1A、m6A、m5C這3種RNA修飾位點進行識別后生成的ROC曲線。從圖2可看出,m1A、m6A、m5C所對應的AUC值分別為0.998 6、0.931 2和0.955 8,表明XGBoost預測模型的魯棒性非常好。
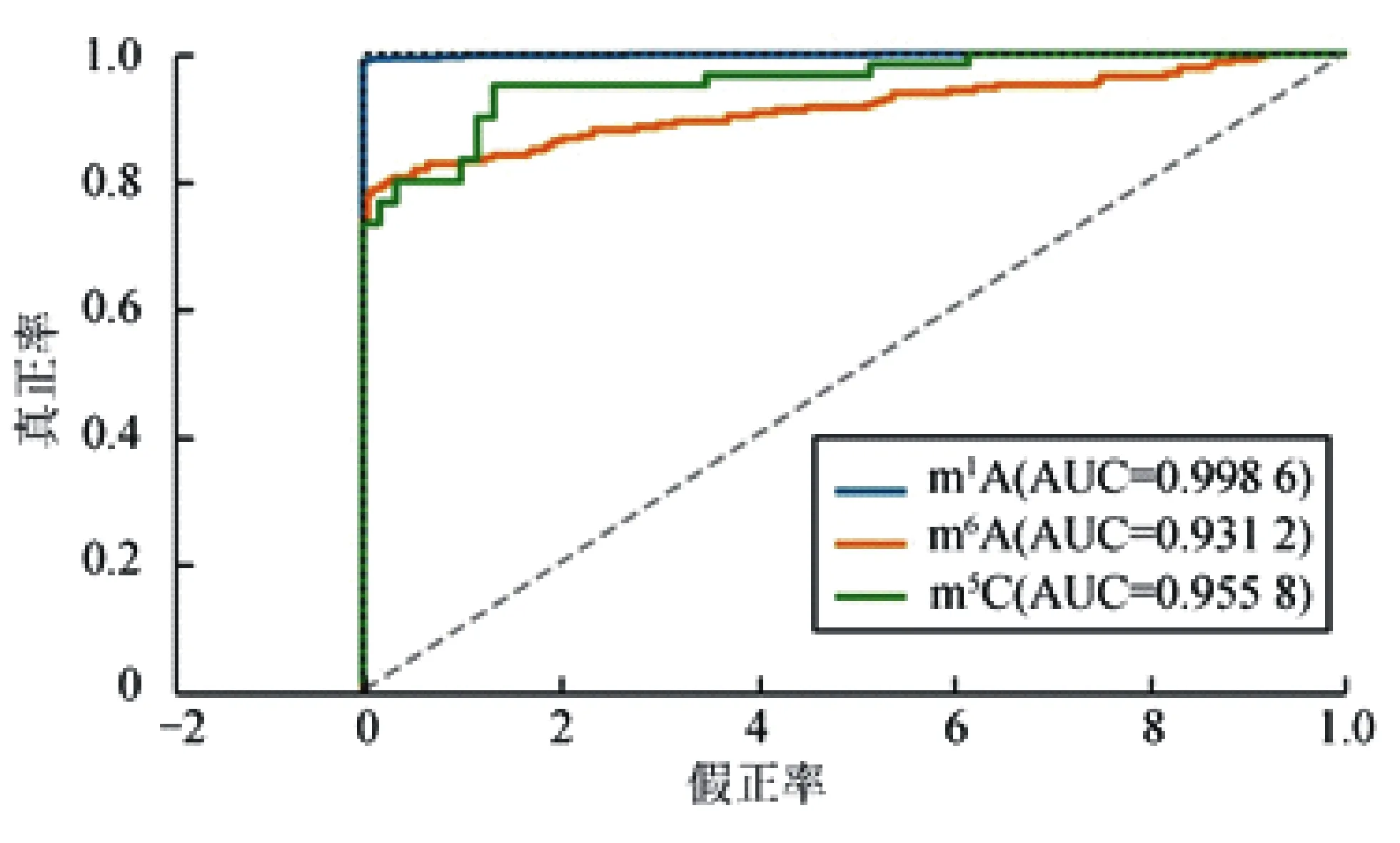
圖2 XGBoost模型在m1A,m6A和m5C修飾位點上的識別性能
2.3 不同方法的識別結果對比
將XGBoost預測模型的識別結果與文獻[13]使用的SVM預測模型的識別結果進行對比,2個模型經過jackknife測試后的結果如表3所示。從表3可看出,XGBoost預測模型和SVM預測模型在m1A修飾位點的識別上均取得了較好的結果,準確率分別達到了99.9%、99.1%;在m6A修飾位點的識別上,XGBoost預測模型的準確率為92.6%,SVM預測模型的準確率為90.4%,提升了2.2%;在m5C修飾位點的識別上,XGBoost預測模型的準確率達到了89.6%,遠高于SVM預測模型的77.5%,提升了12.1%。
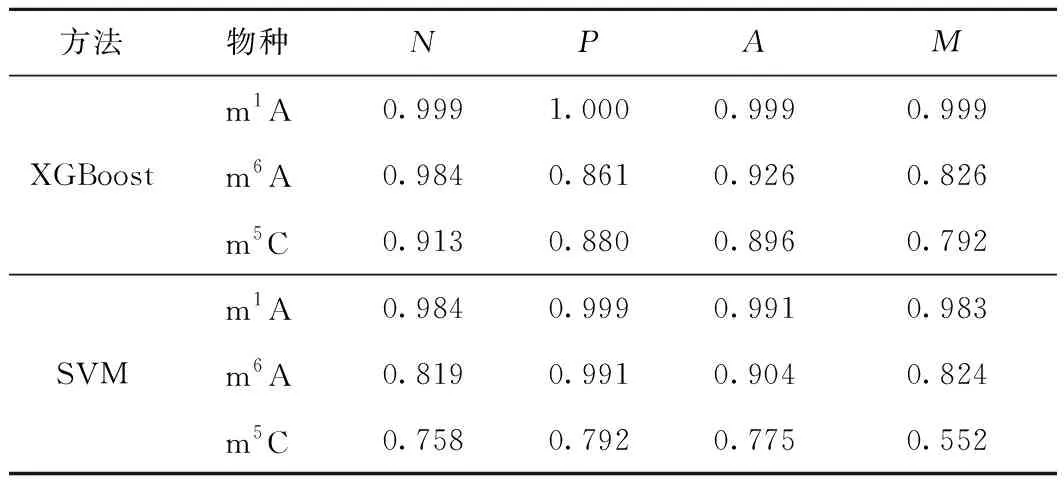
表3 XGBoost與SVM的識別結果比較
3 結束語
為了更快速、準確地識別RNA序列中的修飾位點m1A、m6A、m5C,提出了一種融合位置特異性單核苷酸及雙核苷酸偏好特征的PseKNC編碼方式,并構建了一個基于XGBoost的RNA修飾位點的預測模型。與現有的SVM預測模型相比,其準確率和馬修斯相關系數均取得了明顯提升,其中,對于在現有SVM預測模型上識別效果相對較差的m5C修飾位點,在XGBoost預測模型上取得了較大的突破,識別準確率從77.5%提高到了89.6%,馬修斯相關系數從0.552提高到了0.792,此外,敏感性和特異性也分別從0.758和0.792提高到了0.913和0.880。XGBoost預測模型的提出為RNA修飾位點的識別提供了高效、可靠的方法。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
