CD8+T細胞PD-1基因CRISPR/Cas9編輯體系電穿孔條件的優化
2019-06-03 06:23:02王淑敏張代群
鄭州大學學報(醫學版) 2019年3期
關鍵詞:效率
王淑敏,張代群,李 峰,張 毅
1)鄭州大學第一附屬醫院生物細胞治療中心 鄭州 450052 2)鄭州大學第一附屬醫院腫瘤中心 鄭州 450052 3)鄭州大學生命科學學院 鄭州 450001 4)河南省腫瘤免疫與生物治療重點實驗室 鄭州 450052
目前,免疫治療已經成為最具前景的腫瘤治療手段之一,T細胞在腫瘤免疫治療中起到關鍵作用。然而,在實體瘤腫瘤微環境中存在多種免疫逃逸機制,可以識別T細胞表面的免疫檢查點,使T細胞不能對腫瘤細胞進行有效的識別和殺傷,并且走向耗竭狀態[1],抑制免疫檢查點可以刺激或增強抗腫瘤免疫應答[2]。PD-1存在于活化的T細胞和調節性T細胞(Treg)上,其配體PD-L1可在腫瘤細胞和樹突狀細胞[3-5]上表達。腫瘤細胞表面的PD-L1分子與T細胞表面的PD-1結合,可以抑制T細胞的免疫活性;阻斷兩者的結合,可恢復T細胞的抗腫瘤作用。成簇的規律間隔短回文重復序列(CRISPR)/CRISPR相關蛋白9(Cas9)已經成為時下最流行的可以在多種物種中進行基因編輯的工具[6]。相比于傳統重組鋅指核酸酶(zinc finger nuclease, ZFN)和轉錄激活因子樣效應蛋白核酸酶(transcription activator-like effector nuclease, TALEN)等技術,CRISPR/Cas9技術具有操作簡單、效率高、成本低的優點[7]。利用CRISPR/Cas9對人原代T細胞進行基因編輯,為基于T細胞的免疫治療提供了巨大的應用前景[8],但CRISPR/Cas9編輯體系會對細胞活性造成一定損害。目前最普遍應用的是Lonza公司的4D-Nuleofector儀器,其配有不同的電轉緩沖液和脈沖組合,可針對各種類型的細胞進行電穿孔[9]。本文根據文獻報道和前期研究[9-10],設計兩種電轉緩沖液和脈沖組合方案,檢測兩種方案編輯CD8+T細胞PD-1基因的效率,比較兩種方案對T細胞生物學特性的影響,旨在篩選出一種更適合T細胞基因編輯的電穿孔條件。
1 材料與方法
1.1主要材料與試劑胎牛血清、高糖DMEM培養基、RPMI 1640培養基均購自美國Sigma公司,人外周血淋巴細胞分離液購自天津灝洋生物制品有限公司,CD3單抗、CD28單抗、高保真DNA 聚合酶、Lipofectamine 3000轉染試劑、Cas9核酸酶購自美國Thermo Fisher Scientific公司,T7核酸內切酶1(T7E1)和BbsⅠ酶購自美國NEB公司,流式熒光抗體均購自美國Biolegend公司,重組人IL-2購自北京雙鷺藥業股份有限公司,T7體外轉錄試劑盒購自南京諾唯贊生物科技有限公司,蛋白酶K、質粒提取試劑盒購自天根生化科技(北京)有限公司,RNA純化試劑盒購自美國Zyom Research公司,T4 DNA連接酶、Stabl3感受態細胞、Premix Taq、基因組DNA提取試劑盒和Primer STAR聚合酶均購自日本TaKaRa公司,DNA純化試劑盒購自美國Axygen公司,電轉儀和電轉緩沖液均購自瑞士Lonza公司,質粒pSpCas9(BB)-2A-GFP (PX458)購自美國Addgene公司,人胚腎293T細胞購自中國科學院細胞所。
1.2PD-1sgRNA序列及引物的設計與合成使用在線工具http://crispr.mit.edu/設計3條PD-1 sgRNA序列,使用NCBI網站的Primer-blast在線設計3條PD-1 T7E1檢測引物及PD-1 vitro上下游引物、PX458 check檢測引物。PD-1 T7E1檢測引物設計在編輯位點的上游150~200 bp和下游350~400 bp處(反之亦可)。所有序列均由上海生工生物工程股份有限公司合成,見表1。

表1 sgRNA及引物序列
1.3靶向PD-1重組PX458質粒的構建按圖1模式構建質粒。用BbsⅠ酶線性化PX458質粒,并使用DNA純化試劑盒純化酶切產物,檢測濃度。將合成好的sgRNA單鏈混合并稀釋至1 mol/L,退火形成雙鏈,按照sgRNA∶線性PX458質粒物質的量比為5∶1混合,然后加入1 μL T4 Ligation buffer、1 μL T4 DNA連接酶,終體積為10 μL,16 ℃連接過夜。將連接產物轉化Stabl3感受態細胞,涂布于帶有氨芐青霉素的平板,挑取單個克隆至含有LB肉湯培養基和氨芐青霉素的2 mL離心管中,在37 ℃、250 r/min水平搖床中培養2~3 h,然后做菌液PCR。PCR體系:1.5 μL菌液,1.0 μL PX458 check上游引物,1.0 μL PD-1 sgRNA下游引物,10.0 μL Premix Taq,無菌水6.5 μL。PCR反應條件:95 ℃ 10 min;95 ℃30 s,55 ℃ 30 s,72 ℃ 50 s,共35個循環;72 ℃ 5 min,4 ℃保持。將PCR產物進行凝膠電泳,選擇能擴增出單一且明亮條帶的菌液,轉移至搖菌管中,37 ℃搖床中220 r/min過夜,抽提質粒,送上海生工生物工程有限公司測序驗證。
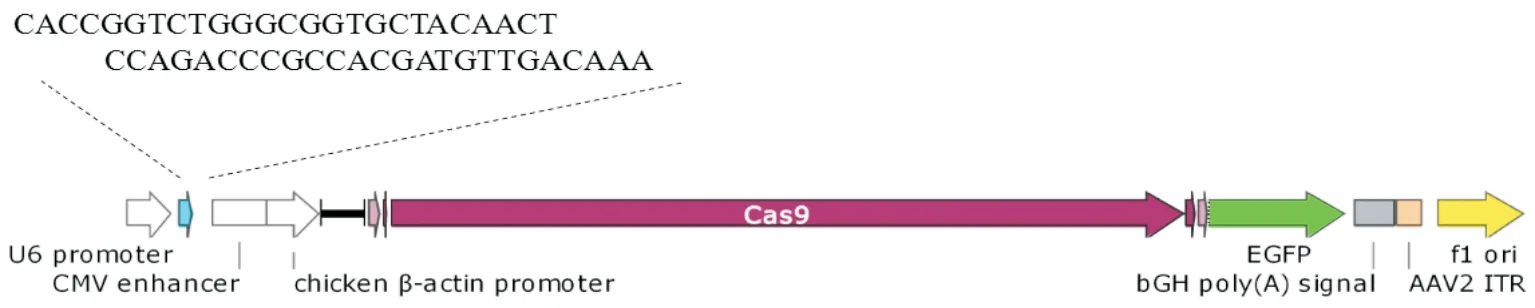
圖1 重組質粒示意圖
1.4293T細胞培養與轉染將處于對數生長期的293T細胞以4×105個/孔的密度接種到6孔板,在含高糖DMEM培養基(含體積分數10%胎牛血清)、體積分數5%CO2、37 ℃恒溫培養箱中培養24 h,待細胞融合度達到70%左右開始轉染。轉染前2 h更換為無血清培養基對細胞進行“饑餓”處理,用Lipofectamine 3000將1.3中構建好的重組質粒轉入293T細胞中,以空PX458質粒為陰性對照。
1.5PD-1切割效率的檢測轉染48 h后,消化293T細胞,用PBS洗3次,按照試劑盒說明書操作提取基因組DNA,使用Primer STAR聚合酶和PD-1 T7E1檢測引物進行PCR,擴增編輯位點的DNA片段。以純化后的PCR產物為模板行PCR。反應體系:DNA片段200 ng,10×NE Buffer 2 μL,無核酸酶水補充至19 μL,行PCR。反應條件:95 ℃ 5 min,95~85 ℃(按2 ℃/s梯度程序降溫至85 ℃),85~25 ℃(按0.1 ℃/s梯度程序降溫至25 ℃)。然后加入1 μL T7E1酶切15 min,立即行20 g/L的瓊脂糖凝膠電泳,用凝膠成像儀拍照觀察。若靶點被CRISPR/Cas9成功編輯,經PD-1 T7E1酶切,電泳結果會呈現3條帶,大小分別約為600、400和200 bp。用Image J軟件對條帶進行灰度分析,計算切割效率。切割效率=(200 bp條帶灰度值+400 bp條帶灰度值)/3條條帶灰度值總和×100%。
1.6sgRNA的體外轉錄及活性驗證以插入PD-1 sgRNA2的重組PX458質粒為模板,加入PD-1 vitro上、下游引物,PCR擴增出體外轉錄模板,用DNA純化試劑盒純化。參照說明書,用T7E1體外轉錄試劑盒將模板轉錄成RNA,用RNA純化試劑盒對sgRNA進行純化,于-80 ℃保存。
以293T細胞基因組為模板,用T7E1檢測引物進行PCR和凝膠電泳,回收產物并純化,以產物為模板行PCR。擴增體系19 μL,包括10×Cas9 Buffer 2 μL體外轉錄的2 μmol/L sgRNA 2 μL,1 μmol/L的Cas9 2 μL,適量去核酸酶水,室溫孵育10 min,模板DNA0.2 pmol,37 ℃孵育30 min之后再加入1 μL蛋白酶K,37 ℃反應15 min,進行瓊脂糖凝膠電泳,用凝膠成像儀拍照,用Image J軟件對條帶進行灰度分析。以切割效率反映sgRNA活性,計算方法同1.5。
1.7CD8+T細胞電穿孔條件外周血單個核細胞的分離、CD8+T細胞的分選、培養和活化見參考文獻[11]。收集活化后的CD8+T細胞,離心棄上清。將3 μg sgRNA和3 μg Cas9室溫(25 ℃)共孵育10min,用電轉染緩沖液稀釋,用此稀釋液重懸CD8+T細胞至3×104個/μL,轉移到100 μL的電轉杯中,在Lonza 4D-Nuleofector儀器中分別用方案一(P2電轉染緩沖液+EH100脈沖)、方案二(P3電轉染緩沖液+EO115脈沖)進行電穿孔。以只加Cas9未加sgRNA的細胞為對照。所用的電轉染緩沖液均為Lonza公司原廠包裝,脈沖均為Lonza 4D-Nuleofector儀器內置程序。電穿孔后的細胞用37 ℃預熱的含有300 IU/mL IL-2、體積分數10%胎牛血清的RPMI 1640培養基,于24孔培養板中在體積分數5% CO2、37 ℃恒溫培養箱中培養。
1.8電穿孔后CD8+T細胞活性及PD-1敲除效率、分化狀態和增殖能力的檢測以未電穿孔的細胞為對照組,分別用1.8項兩種方案(方案一組、方案二組)對活化3 d的CD8+T細胞進行電穿孔,以敲除PD-1。①繼續培養48 h后收取細胞,加入7AAD-PerCP流式抗體,檢測細胞活性。②電穿孔后的細胞培養3 d后,再次加入CD3單抗、CD28單抗刺激6 h,加入抗人CD8-APC-Cy7、PD-1-FITC、7AAD-PerCP流式抗體,在4 ℃條件下避光孵育15 min,上流式細胞儀檢測。PD-1敲除效率=(對照組PD-1表達量-實驗組PD-1表達量)/對照組PD-1表達量×100%。③分別取3組CD8+T細胞1×105個/管,加入CD8-APC-Cy7、CD45RO-APC、CCR7-PE,4 ℃避光孵育15 min后,上流式細胞儀檢測。CD45RO-CCR7+為初始T細胞(Tn),CD45RO+CCR7+為中心記憶性T細胞(Tcm),CD45RO-CCR7-為效應T細胞(Teff),CD45RO+CCR7-為效應記憶性T細胞(Tem)。④分別將3組CD8+T細胞按照5×106個/孔鋪于12孔板中,置于37 ℃、體積分數5%CO2培養箱中培養。第2、4、6和8天收取細胞,混勻后吸取10 μL于血球計數板計數,計算增殖倍數。增殖倍數=終細胞數/初始細胞數。
1.9統計學處理采用SPSS 21.0進行分析,應用單因素方差分析和LSD-t檢驗分析3組CD8+T細胞活性、細胞增殖倍數的差異,應用t檢驗比較兩種方案PD-1敲除效率的差異,檢驗水準α=0.05 。
2 結果
2.1靶向PD-1的PX458重組質粒測序結果及PD-1抑制效率測序結果見圖2。T7E1核酸內切酶法驗證果見圖3。由圖3可知,sgRNA1、2、3介導的PD-1基因切割效率分別為60.32%、68.61%、40.96%,其中PD-1 sgRNA2切割效率最高。體外轉錄sgRNA2對293T細胞PD-1基因的切割效率達93%(圖4)。
2.2方案一、二組PD-1敲除效率的比較結果見圖5。方案一組敲除效率[(87.70±1.03)%]明顯高于方案二組[(70.13±2.13)%](t=7.440,P=0.002)。
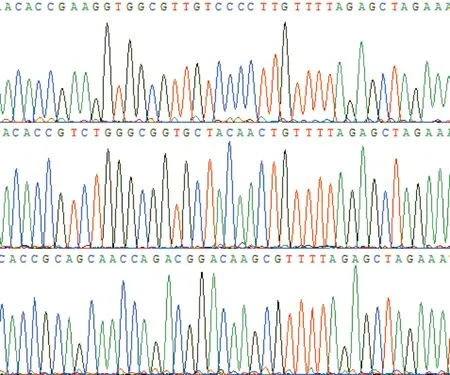
上、中、下:PD-1-sgRNA1-PX548、PD-1-sgRNA2-PX548和PD-1-sgRNA3-PX548測序結果
圖2測序結果
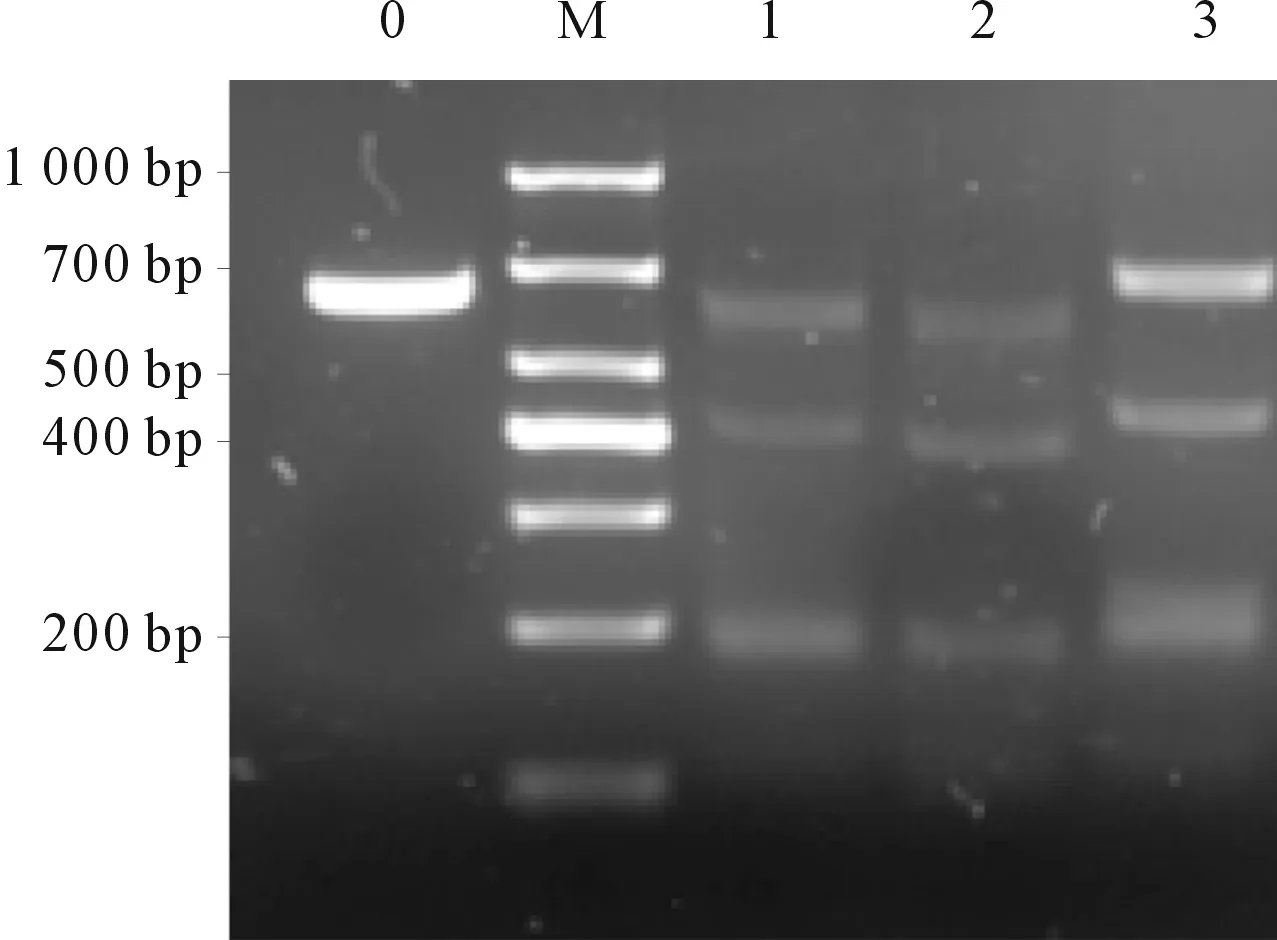
M:Marker;0:陰性對照;1、2、3:PD-1 sgRNA1、2、3
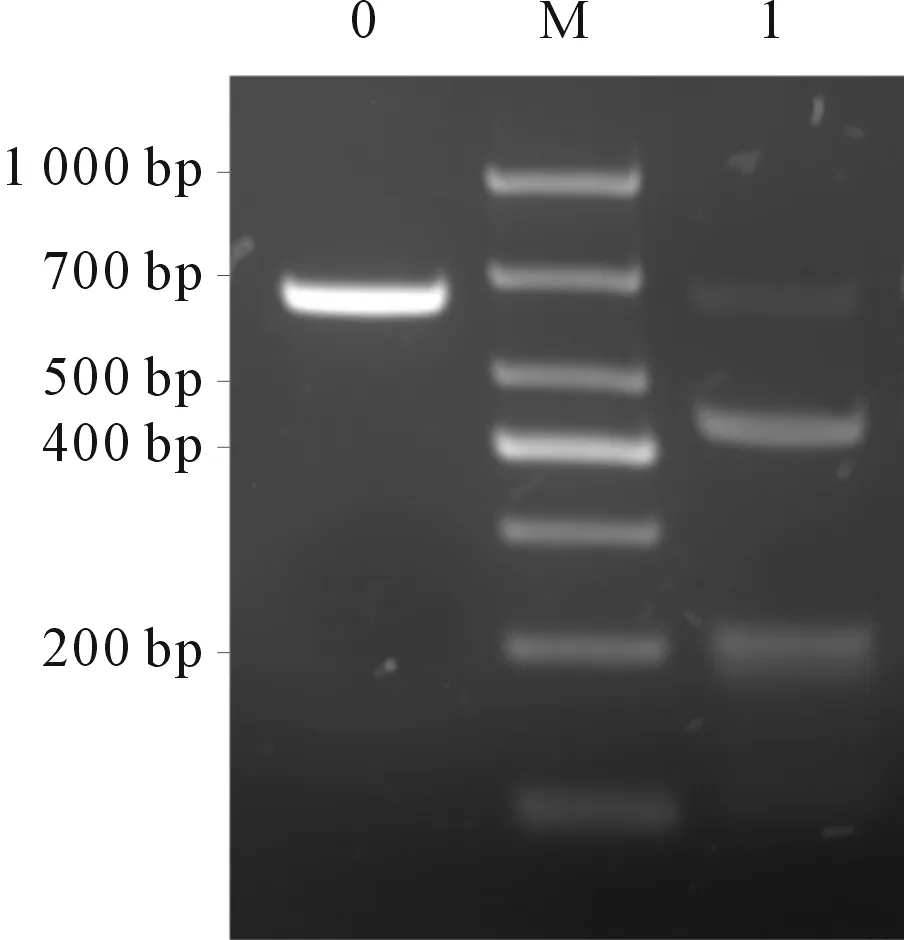
M:Marker;0:陰性對照;1:PD-1 sgRNA2
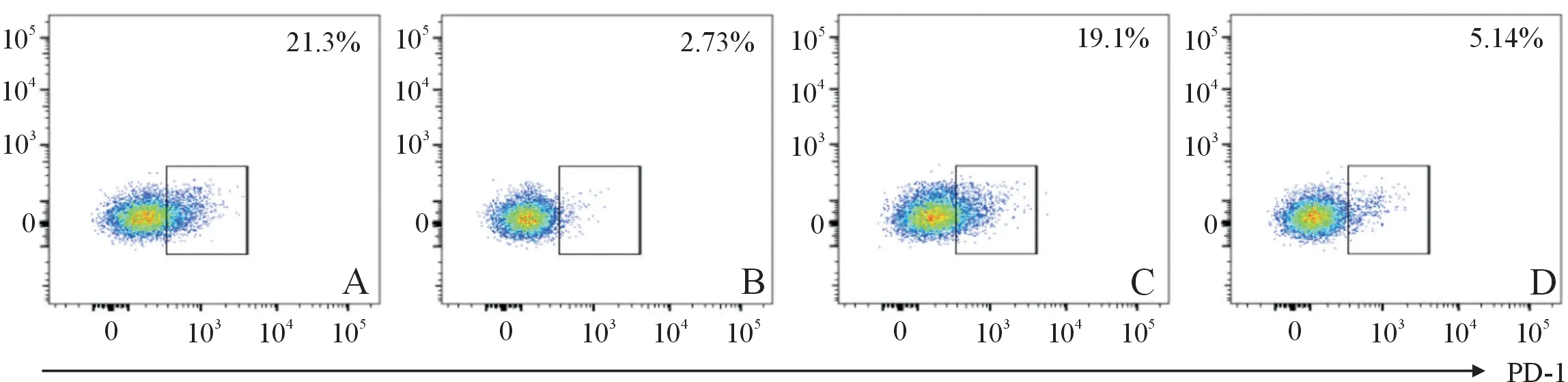
A:方案一對照組;B:方案一實驗組;C:方案二對照組;D:方案二實驗組
2.3電穿孔后CD8+T細胞活性結果見圖6。對照組[(1.07±0.15)%]、方案一組[(1.24±0.13)%]和方案二組[(1.56±0.28)%]死細胞比例差異無統計學意義(F=4.657,P=0.060)。
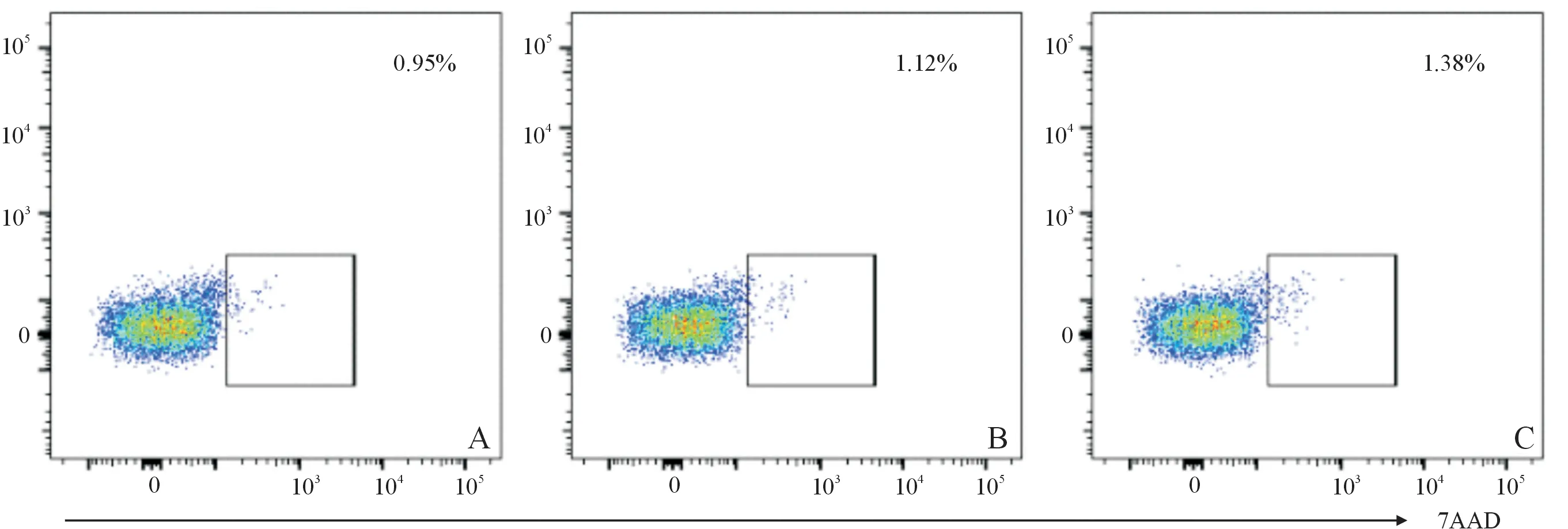
A:對照組;B:方案一組;C:方案二組
2.43組CD8+T細胞分化狀態的比較見圖7、表2。3組CD8+T細胞的分化狀態差異無統計學意義。
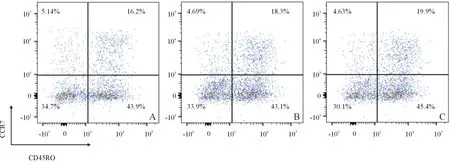
A:對照組;B:方案一組;C:方案二組
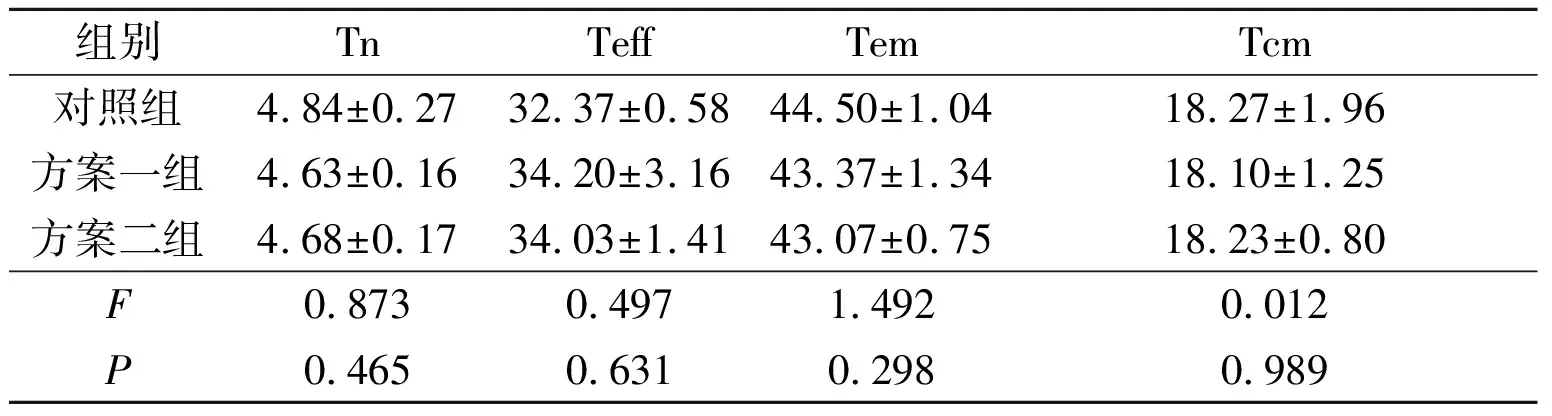
表2 3組CD8+T細胞分化狀態的比較(n=3) %
2.53組CD8+T細胞增殖倍數的比較見表3。由表3可知,3組細胞增殖倍數差異無統計學意義。
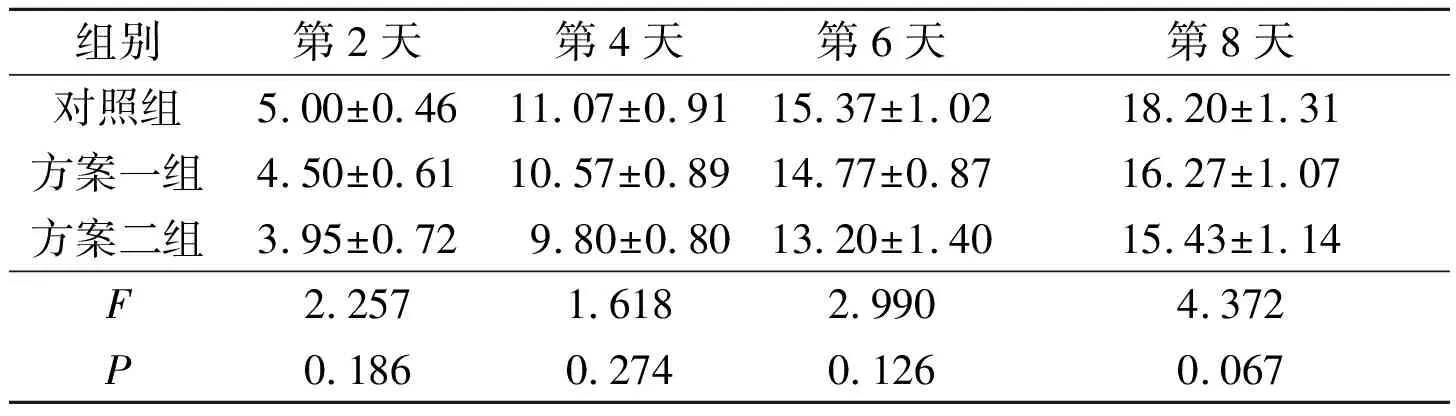
表3 3組CD8+T細胞增殖倍數的比較(n=3)
3 討論
T細胞可以通過抗原提呈細胞特異性識別腫瘤細胞表面抗原,并有效地殺傷腫瘤細胞,然而,腫瘤微環境中存在復雜的機制,使抗腫瘤免疫反應延遲、改變甚至停止[12],即“免疫逃逸機制”。免疫逃逸導致自身免疫系統不能有效識別并殺傷腫瘤細胞,導致腫瘤生長失去控制而處于異常過度增殖狀態[13]。通過抑制這些免疫檢查點來解除患者免疫抑制狀態,增強抗腫瘤免疫反應,目前被認為是最有前景的腫瘤治療方法之一。
已有文獻[14-17]報道使用Cas9蛋白和體外轉錄或合成的sgRNA形成的復合物進行電穿孔可以有效地對人原代T細胞進行基因編輯。CRISPR/Cas9作為一種基因編輯技術,目前被廣泛應用,該技術具有許多明顯的優勢:沒有物種限制;基因編輯方式多樣(敲除、插入[18]、抑制、激活等);可實現對多個靶基因同時編輯;操作較簡單,且編輯效率高[12]。利用該技術對T細胞進行基因編輯,如敲除一個或多個免疫檢查點,之后進行過繼回輸,可以減少免疫抑制的發生,增強T細胞的抗腫瘤免疫反應[8]。然而向T細胞中遞送CRISPR/Cas9系統的方法有很多種,但基因編輯效率及基因編輯后對T細胞活性的影響還有待優化。因此本研究采用Lonza公司的4D-Nuleofector儀器遞送靶向T細胞PD-1的CRISPR/Cas9蛋白,根據已有文獻報道和本實驗室前期實驗,選擇兩種該儀器配有的電轉染緩沖液和脈沖組合進行對比,即方案一(P2電轉染緩沖液 + EH100脈沖)與方案二(P3電轉染緩沖液 + EO115脈沖),通過檢測細胞上PD-1基因編輯效率及編輯后細胞活性、分化與增殖,鑒定出P2電轉染緩沖液和EH100脈沖的組合是一種更加高效的電穿孔條件,可以有效地提高T細胞基因編輯效率并且不會影響T細胞的活性、分化和增殖。
猜你喜歡
瘋狂英語·初中天地(2021年5期)2021-07-21 02:24:28
甘肅教育(2020年14期)2020-09-11 07:57:42
中學生數理化(高中版.高考數學)(2020年5期)2020-06-02 09:19:08
商周刊(2017年9期)2017-08-22 02:57:49
遼寧經濟(2017年6期)2017-07-12 09:27:16
中國衛生(2016年9期)2016-11-12 13:27:54
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國洗滌用品工業(2015年7期)2015-02-28 19:02:38
電子設計工程(2015年12期)2015-02-27 12:06:10
中國衛生(2014年11期)2014-11-12 13:11:32
