基因編輯技術原理及其在動植物研究中的應用
2019-06-11 00:39:56張泗舉欒維江
天津師范大學學報(自然科學版) 2019年3期
關鍵詞:系統
張泗舉,欒維江
(1.天津師范大學生命科學學院,天津 300387;2.天津師范大學天津市動植物抗性重點實驗室,天津 300387)
自從人們認識基因的本質是DNA 以來,就嘗試著采用各種方法對基因進行一系列的改變,以期通過創造相應的突變體,獲得對人類有益的微生物和動植物性狀或應用于人類的疾病治療.在這個過程中,人們運用了物理方法、化學方法、分子生物學方法等,對基因的改變也從當初的不定向突變發展為定向改變.在定向改變技術中,又經歷了ZFN、TALEN、CRISPR/Cas 等技術的發展過程.本文對這一過程進行了系統梳理,對各種方法的原理及應用范圍進行了簡要總結,并著重介紹以CRISPR/Cas9 為代表的基因編輯技術的發展歷程、基本原理、應用范圍及注意事項等.此外,對基因編輯的前景作了展望.
1 非定向誘變技術
1.1 理化誘變技術
非定向打靶技術主要包括物理方法和化學方法.物理方法主要通過各種射線來處理生物材料,造成生物體DNA 的斷裂或交聯等損傷.常用的射線有X 射線、γ 射線、中子、電子束、紫外線等.輻射可產生A-T或C-G 之間的氫鍵斷裂、 在1 或2 個DNA 鏈中糖與磷酸基之間發生斷裂、同一DNA 上相鄰胸腺嘧啶之間形成二聚體以及DNA 鏈的斷裂和交聯等多種結果[1].這些損傷如果得不到正確修復,就會產生突變.化學方法主要是使用能引起DNA 序列改變的化學試劑,包括:①烷化劑類,它們能置換DNA 分子的H 原子(烷化作用),改變基因的分子結構;②核酸堿基類似物,在不妨礙DNA 復制的情況下,代替DNA 的成分滲入到DNA 分子中去,引起DNA 復制時堿基配對的差錯;③吖啶類嵌入劑,誘發移碼突變; ④亞硝酸,能使核酸、核苷酸和核苷中的嘌呤和嘧啶上的氨基轉變為羥基,造成DNA 復制的紊亂[2].
理化誘變僅造成個別或者一些位點的DNA 結構發生變化,總體的遺傳背景是一致的,因此在生物學研究特別是基因克隆和功能研究中該技術受到高度重視.理化誘變方法簡便,突變效率高,突變由DNA點突變、缺失、重排引起,已廣泛應用于擬南芥和水稻突變體庫的構建中.但其誘變過程難以控制,一個突變體經常包含多個點突變,突變表型可能由多個點突變引起,增加了基因功能鑒定的難度.
1.2 DNA插入突變技術
插入突變是T-DNA(Transfer DNA)、轉座子標簽(Transposon tagging)或逆轉座子標簽(Retrotransposon tagging)插入到基因組中,相應位點基因的功能可能受到抑制而產生基因敲除(Knock out)突變體,插入元件同時又可用作標簽從基因組中分離出相應位點的基因并鑒定其功能.T-DNA、逆轉座子標簽和轉座子標簽是構建插入突變體庫的3 種主要方法,經常用于模式植物的突變體創建[3].農桿菌介導的植物遺傳轉化方法的成熟和完善為插入突變體庫的構建提供了依據[4].
T-DNA插入突變是農桿菌侵染植物后,農桿菌Ti 質粒中的一段DNA 整合到植物基因組中.如果插入位點是功能基因,通常會造成該基因的功能喪失,如果該基因是植物生長發育不可缺少的,則會表現出相應的突變性狀.T-DNA 在外源植物基因組中的整合是隨機的,它可以整合到植物的任何一條染色體上.T-DNA 插入的位置不同,可引起植物產生不同表型.利用T-DNA 方法構建插入突變體庫,每個轉基因植株的產生都是獨立的遺傳轉化事件,是發現基因突變的有效手段.但轉基因步驟復雜,有些植物的轉化效率不高,因此構建T-DNA 插入的大群體費錢費功,而且伴有大量的逆轉座子轉座事件,提高了研究難度.T-DNA插入突變方法多應用于水稻、 擬南芥等植物[4-5].
轉座子插入突變包括2 種類型: 一種是以DNA介導的轉座子,通過切粘機制復制并發生轉座,如玉米Ac/Ds 系統、En/Spm 系統、金魚草Tam3 等[6].不同于T-DNA 標簽法需要成千上萬次的遺傳轉化事件才能構建大規模的插入突變體庫,這種轉座子系統可以通過轉座子的轉座活性在基因組中轉座而產生突變體,構建插入突變體庫.它既具有T-DNA插入突變體庫信息量大的優點,又不需要數以十萬計的轉基因植株,大大減少了轉基因的工作量.另一種是逆轉座子插入突變,它是以RNA 介導的轉座方式,與DNA 介導的轉座子不同,它可以通過反轉錄后經復制產生一個新的拷貝插入到基因中,而原來的拷貝仍然存在.目前在水稻中應用較多的是水稻內源逆轉座子Tos17,研究發現,組織培養可以使得Tos17 轉座子變得活躍,培養時間越長,拷貝數變得越多[7].因而該方法被廣泛應用于水稻突變體庫的構建,通過對突變體進行分析,鑒定了一些重要的功能基因[7-8].
2 定向打靶技術
人類基因組計劃開展以來,基因組測序技術得到了飛躍式的發展,越來越多生物的基因組得到了測定與解析.除了人類基因組,重要的模式生物(如線蟲、酵母、小鼠、擬南芥)、農作物(如水稻、玉米、大豆)以及牲畜(如豬、牛)等生物的基因組都得到了測定.在此基礎上,人們希望對生物的單個基因進行精確操作以期研究基因的功能,進而控制基因的表達,獲得有益的生物性狀.在這種需求下,人們逐步發展了以下幾種基因定向操作技術.
2.1 ZFN編輯技術
鋅指核酸酶(Zinc-finger nucleases,ZFNs)技術是將具有鋅指結構且能夠識別特定堿基序列的多肽與Ⅱ型核酸酶的FokⅠ結構域融合表達,分別結合于互補雙鏈的2 個融合蛋白形成二聚體,對DNA 雙鏈進行切割[9].該技術的構思最早來源于人們對小鼠鋅指蛋白Zif268 結構的解析,Pavletich 等[10]發現小鼠Zif268 蛋白共有90 個氨基酸,每30 個氨基酸構成一個鋅指單體,一個鋅指單體可識別3個相鄰的堿基.將多個鋅指單體串聯后,就可以結合多個相鄰的堿基,與Ⅱ型核酸酶的切割結構域融合后,形成二聚體,即可切割DNA 序列,產生雙鏈DNA 斷點(Double strand break,DSB).由于斷點的產生,生物就會啟動自身修復系統進行斷點的修復,主要通過同源重組(Homologydirected recombination,HDR)和非同源末端連接(Nonhomologous end joining,NHEJ)2 種方式修復.無論哪種修復方式,都會造成在斷點附近的DNA 插入或缺失,從而引入突變.該技術主要在人類細胞、 煙草、斑馬魚、果蠅、線蟲等模式生物中得到應用[11].ZFN 技術是最早被應用于基因定向打靶的技術,由于三聯體識別序列的種類較少、技術存在專利保護以及當時測序的物種較少等原因,該技術的應用并不十分廣泛.
2.2 TALEN編輯技術
TALEN,即轉錄激活子樣效應因子核酸酶(Transcription activator-like effector nucleases,TALENs),其構建思路與ZFN 相似,將轉錄激活子樣效應因子(TALE)與Ⅱ型核酸酶的FokⅠ結構域融合表達,2 個不同的融合蛋白分別結合互補雙鏈后形成二聚體,對DNA 雙鏈進行切割,產生DSB,然后又用上述方式進行修復產生突變.
TALE 是從黃單胞菌屬細菌中發現的蛋白質類的毒力因子,在侵染植物時,該因子通過Ⅲ型分泌途徑注入到植物的細胞質中,然后在核定位信號的引導下進入細胞核,與核DNA 結合而激活寄主細胞的基因轉錄[12].TALE 的結構分為3 部分,即中心串聯重復結構域、核定位信號區(NLS)和酸性轉錄激活區(AAD).中心串聯重復結構域是識別并結合DNA 的位點,由多個TALE 單體組成,單體的個數從1.5~28.5 不等,每個單體由34~35 個氨基酸組成[13].
TALE 單體的34 個氨基酸中,只有第12 和第13個氨基酸是重復可變雙氨基酸(Repeat variant di-residue,RVD),決定了單體所識別的4種DNA 堿基的類型,如NI 識別A 堿基、NG 識別T 堿基、HD 識別C 堿基、NN 識別G 或A 堿基[14-15].根據對應關系,可以人為設計TALE 各單體的組成,對特定基因的DNA 序列進行識別,然后用和TALE 融合的核酸酶對DNA 雙鏈進行切割,產生DSB.對TALE 的利用分為2 類:第一類將TALE 與轉錄激活結構域融合,形成TALE-TF,通過激活轉錄,提高基因的表達水平,這在人類全能細胞系和擬南芥中得到了驗證[16-17]; 第二類將TALE與Ⅱ型核酸內切酶的催化結構域FokⅠ融合表達,形成TALEN,TALEN 二聚體切割DNA 雙鏈,產生突變,達到基因功能敲除目的.明尼蘇達大學的研究人員首先將TALE 與FokⅠ融合,并在酵母中進行表達,驗證了其活性[18].之后TALEN 的功能在人類細胞系、斑馬魚、大鼠、線蟲等模式動物中得到了驗證[19-20];在煙草、擬南芥、水稻、小麥等植物中得到了驗證[21-22].
2.3 CRISPR/Cas9編輯技術
2012年之后,一種新型的基因編輯技術—CRISPR/Cas9 技術被廣泛應用于不同物種的基因靶向編輯,該技術同ZFN 及TALEN 技術一樣,都用特異性核酸酶對特定基因DNA 序列進行編輯,但其操作簡單方便,效率相對較高,已廣泛應用于基因功能研究中,對生命科學的發展起到較大的推動作用.
2.3.1 CRISPR 序列的發現及命名
CRISPR/Cas9技術的發現及命名源于一段DNA序列.早在1987年,日本科學家Ishino 等[23]在大腸桿菌中克隆了一個編碼堿性磷酸酶的基因ipa,為了分析該基因的編碼框及蛋白產物,對1.7 kb 的克隆片段進行測序,分析發現1.7 kb 序列包括1 038 bp 的ORF、331 bp 的5’端側翼以及約300 bp 的3’端側翼序列.進一步分析3’端側翼序列發現,約300 bp 的序列中有5 個29 bp 的回文重復序列,并被4 個32 bp的間隔序列隔開.4 個32 bp 的間隔序列各不相同,而且與大腸桿菌的序列沒有同源性.由于局限于當時測序還沒有規模化發展,數據庫中序列比較少,因此作者比對后在其他原核生物中也沒發現其同源序列,也不知道其重要意義何在,作者在以后的研究中未對這段有趣的序列進行深究,直到約10年后與這段序列有相似特點的序列才再次進入科學家的視野.
1990年代初,Mojica 等[24]利用一種耐高鹽的嗜鹽古菌(Haloferax mediterranei)研究鹽濃度如何影響限制性內切酶對其基因組DNA 序列的切割時發現了類似1987年Ishino 等發現的特殊序列,這種特殊序列由多個高度保守的30 bp 的回文序列組成,這些回文序列被多個不相關的36 bp 的非嗜鹽古菌自身的序列居間隔開.后來,Mojica 等[25]又在其他細菌中發現了類似結構的序列,通過比對,發現雖然每種細菌的回文重復及居間序列都不同,但都具有相同的“重復-居間序列-反向重復”的結構,他開始認識到了這種特殊結構序列的重要性.由于1989年人類基因組計劃的實施,測序方法有了長足進展,測序費用也有所降低,因此不同物種的測序計劃也廣泛實施,數據庫中已知的DNA 序列日益增多.為了揭開這種特殊結構序列的生物學意義,Mojica 等[26]進一步利用生物信息學對自己所獲得的序列進行比對搜尋,以期能在不同細菌中找到更多的這種特殊的回文重復序列.截止2000年,他已在20 種不同的微生物中找到這種特殊結構的序列,并命名為短的規律性的間隔重復(Short regularly spaced repeat,SRSR).2年后,科學家們進一步從不同原核生物中發現了更多的這種特殊序列,并根據序列特點進一步規范其命名,將Mojica 命名的SRSR 改為成簇的、有規律間隔排列的短回文重復序列(Clustered regularly interspaced short palindromic repeats,CRISPR)[27].
2.3.2 CRISPR 序列生物學意義的探索
隨著CRISPR 序列的增多,越來越多的科學家被這種特殊的序列所吸引,科學家們認識到它一定具有重要的生物學意義.為了揭開CRISPR 序列的生物學功能,2003年,Mojica 首先改變了研究思路,從以前著重比對搜尋回文序列轉移到著重比對搜尋居間序列,希望能找到回文序列之間的居間序列來自于哪里.通過他的努力,終于在P1 噬菌體中發現與居間序列完全匹配的序列,通過進一步擴大比對居間序列的數目,發現很多居間序列匹配到不同的病毒及外來的質粒中.至此,他認識到CRISPR 序列可能在細菌對抗外來物質的侵染中發揮重要作用[28].同時,來自法國的科學家Vergnaud 和他的研究團隊在鼠疫桿菌(Yersinia pestis)中發現了CRISPR 序列,并揭示出其居間序列來自于噬菌體,對于鼠疫桿菌抵抗噬菌體的侵染具有重要作用[29].來自法國的另外一個研究團隊在嗜熱鏈球菌(Streptococcus thermophilus)中克隆了4 個基因Cas1~Cas4,并在這些基因附近發現了CRISPR 序列,分析這些CRISPR 序列中的居間序列發現它們來自于噬菌體及染色體外遺傳元件(Extrachromosomal elements),進一步的噬菌體敏感性實驗研究發現,這些居間序列對嗜熱鏈球菌抵抗外來侵染物具有重要作用,可能起到細胞免疫作用[30].從發表年份上可以看出,基因編輯技術的研究存在激烈競爭.
2.3.3 CRISPR/Cas 系統作用機制的解析
在弄清楚了CRISPR 序列可能與細菌的免疫有關后,科學家們開始著手解析微生物是如何利用CRISPR序列進行免疫作用的.早在2002年美國國家生物信息研究中心Eugene Koonin 研究組[31]認為,CRISPR 可能與DNA 的修復有關,但是后來擯棄了這種假設.Makarova 等[32]通過對多種細菌中CRISPR 和Cas 序列的基因組比較分析,認為CRISPR/Cas 系統是一種防御機制,用來對抗入侵的噬菌體和質粒,其作用機制可能與真核生物的RNAi 類似.但是將該系統的成分與RNAi 機制相關的蛋白,如雙鏈RNA 特異的解旋酶(dicer)、切割靶標mRNA 的內切酶(slicer)以及RNA依賴的RNA 聚合酶,進行比對后,卻找不到與之匹配的成分.根據CRISPR 中的獨特居間序列與噬菌體和質粒片段同源的事實,Makarova 等[32]提出CRISPR/Cas可作為原核中的siRNA 起作用(psiRNA),通過與靶標mRNA堿基配對,促使其降解或翻譯終止,并推測這個系統包含將外源基因片段整合到自身染色體上以產生對相應成分的遺傳免疫等步驟.按照該假說,CRISPR 序列首先被轉錄成原初RNA 前體,之后進一步剪切變成成熟的siRNA 起作用,但變成成熟siRNA的酶是什么、如何切割成熟仍然未知.
在此假設基礎上,法國微生物學家Barrangou 等[33]證實了居間序列與相應噬菌體之間的對應關系.研究者以乳制品生產中的工程菌嗜熱鏈球菌為對象,用2種基因組序列有93%一致性的近緣噬菌體858 和2972 進行侵染,得到了一些對之不敏感的嗜熱鏈球菌株.測序發現,抗性突變體菌株中含有噬菌體來源的居間序列,當居間序列與噬菌體基因組DNA 存在單堿基多態性(SNP)時,即居間序列突變與噬菌體基因組DNA 序列不一致時,則抗性喪失.細菌中CRISPR 位點整合的噬菌體來源的居間序列越多,對噬菌體的侵染越不敏感.對于已經獲得噬菌體抗性的菌株,將居間序列刪除后,抗性即喪失,將居間序列替換后,也改變了其抗性,這說明居間序列與細菌獲得的抗性具有緊密的對應關系.同時,Barrangou 等[33]還研究了與CRISPR 序列相聯系的Cas 基因與居間序列的關系.改變Cas 基因與間隔序列之間的距離,則抗性也會喪失.抑制Cas5 的轉錄,抗性喪失,抑制Cas7 的轉錄,抗性不受影響,但刪除Cas7 序列,則抗性喪失,這可能是因為Cas7 參與了新的間隔序列的插入.這些實驗進一步表明了與噬菌體序列一致的居間序列的存在,為菌株提供了抗性.Mojica 等[34]通過對多種細菌的多個CRISPR 位點進行比較,發現居間序列所對應的噬菌體或質粒上有一個通用的NGG 結構,該通用結構對于Cas 蛋白識別特異居間靶向序列具有重要作用.
為了進一步揭開CRISPR 序列的作用機制,科學家們開始研究與CRISPR 序列相聯系的Cas 蛋白.荷蘭Wageningen 大學的van der Oost 研究組以大腸桿菌K12 菌株為材料,研究了Cas 基因的功能[35].他們從K12中得到8 個編碼Cas 的基因,分別編碼Cas3(1 個預測的HD 核酸酶融合1 個DEAD 螺旋酶)、CasA、CasB、CasC、CasD、CasE、Cas1(預測的整合酶)和Cas2(核糖核酸內切酶).通過對Cas 蛋白進行標記后純化,發現了由CasA、CasB、CasC、CasD、CasE 5 個蛋白組成的復合體,命名為Cascade.以單鏈spacer 為探針進行northern雜交,發現了一個57 nt 的非編碼RNA 產物,命名為CRISPR RNA(crRNA).進一步研究發現,CasE 是催化pre-crRNA 為成熟的crRNA 所必需的蛋白.CasE 的晶體結構顯示,它包括2 個結構域與1 個類鐵氧化還原蛋白折疊,與其他的RNA 結合蛋白具有高度的結構相似性.點突變實驗表明,CasEH20A喪失了切割活性.他們進一步研究裝載了crRNA 的Cascade 能否產生對λ 噬菌體的抗性,發現Cascade 在Cas3 存在的時候才起作用,并且pre-crRNA 以DNA 為模板時效率更高.5 個Cas 蛋白組成的復合體在對pre-crRNA 到crRNA 的成熟過程中起作用,crRNA 的兩端側翼序列都是重復結構中的序列,后者可能是Cascade 亞基結合的保守位點.CrRNA 引導復合體靶向噬菌體的核酸,由于與靶向的方向無關,沒有極性,因而認為靶標是DNA.之后多個研究表明,Cas6、Csy4 等核糖核酸內切酶類Cas 蛋白切割pre-crRNA 的重復序列產生crRNA[36-37].以上研究確切表明了Cas 蛋白復合體是CRISPR 序列成熟變成小的功能crRNA 的核酸酶,而且它可以靶向外源DNA,對外源DNA 進行切割.至此,人們對CRISPR 的作用機制有了一個較為明確的認識.
隨著CRISPR 作用機制的解開,科學家們發現現有的CRISPR 加工系統(需要較多的Cas 蛋白復合體)非常復雜,不利于CRISPR 的應用,因此希望找到較為簡單的CRISPR 系統以便于應用.Deltcheva 等[38]發現,在一些細菌的CRISPR/Cas 中,缺少加工crRNA的核糖核酸內切酶(CasE,Cas6).研究者以人源致病菌化膿性鏈球菌(Streptococcus pyogenes)為研究對象,通過差異化RNA 測序發現了反式編碼的小RNA,它有24nt 與CRISPR 前體RNA 中的重復序列互補,稱之為tracrRNA(trans-crRNA).他們在化膿性鏈球菌中發現了與CRISPR 序列相聯系的Csn1(后來命名為Cas9)蛋白,揭示出tracrRNA 是通過廣泛保守的RNaseⅢ和Csn1 蛋白指導crRNA 的成熟,所有這些成分都是防御所必需的.Deltcheva 發現的CRISPR/Cas 系統就是后來被廣泛使用的CRISPR/Cas9 系統.Nature 雜志專門配發了評論文章[39],該評論指出,Csn1 指導的crRNA的成熟過程與其他幾種機制完全不同.這一系統成分簡單,便于操作,為后來CRISPR/Cas9 系統的廣泛應用起到理論指導作用.
為了方便CRISPR/Cas 系統的應用,來自多個國家和實驗室的研究人員一起協作,對CRISPR-Cas 系統的進化關系和分類進行了系統論述[40].CRISPR-Cas系統的主要元件是Cas 操縱子,被分布其上成簇的重復序列居間隔開.CRISPR/Cas 免疫過程大致分為3個階段:第1 個是適應階段,當有外源病毒或質粒入侵時,Cas 操縱子捕獲帶有PAM(Proto-spacer adjacent motif,序列為NGG)結構特征的外源DNA 片段,整合到操縱子的重復序列之間;第2 個是表達階段,整合了外源DNA 片段的Cas 操縱子轉錄為RNA,與相應的Cas 蛋白形成復合體并切割為較短的crRNA,crRNA中含有外源片段及重復序列;第3個是干擾階段,在crRNA 的引導下,Cas 蛋白將與crRNA 中外源片段同源的雙鏈DNA 切割,達到防御目的.根據crRNA 加工的途徑和必要的Cas 蛋白的數目,CRISPR/Cas 系統分為3 類.其中第1 類和第3 類crRNA 需要裝載到多個蛋白構成的復合體中,不方便工程化;第2 類只需要一個Cas9 蛋白起作用,利用宿主的RNaseⅢ使crRNA 成熟.Cas9 也可以單獨起切割作用,通過位點突變分析發現,免疫作用依賴于Cas9 蛋白的HNH 結構域和RuvC 結構域.
3 CRISPR/Cas9編輯技術的應用
3.1 CRISPR/Cas9編輯技術在動物研究中的應用
在CRISPR/Cas 系統的作用機制被揭示清楚后,科學家們認識到它可能具有巨大的應用前景,基于以前ZFN及TALEN編輯技術的應用,CRISPR/Cas 系統也可能在其他的真核生物中工作.為了驗證該系統是否可以編輯其他生物的基因組DNA 序列,霍華德休斯醫學研究所(HHMI)的Doudna 課題組和瑞典于默奧大學的Charpentier 課題組合作,最早將CRISPR/Cas技術進行實際應用[41].研究者將純化的Cas9 蛋白與crRNA 和tracrRNA 混合,在體外實現了對質粒DNA或線性雙鏈DNA 的有效切割.他們首先解析了Cas9蛋白的結構,表明該蛋白約由1 367 個氨基酸組成(不同菌株有差異),N 端的Ruvc I 結構域和C 端的HNH 結構域是其催化結構域.Cas9:: gRNA(引導RNA)復合體結合靶標DNA 雙鏈后,Ruvc I 結構域切割gRNA 的非互補鏈,HNH 結構域切割互補鏈,產生雙鏈斷裂.將Cas9 蛋白第10 位的天冬氨酸突變為丙氨酸,Ruvc I 結構域喪失活性;將第840 位的組氨酸突變為丙氨酸,HNH 結構域失去功能.使用任一結構域突變的Cas9 蛋白,可導致靶標DNA 雙鏈產生切刻(nick)而非斷裂,突變的Cas9 蛋白可用于對純合致死的基因進行編輯.基于此,他們簡化了該系統的作用機制,將crRNA 和tracrRNA 融合表達,形成gRNA,再將Cas9 蛋白和gRNA 分別表達融合,這樣就繞過了crRNA 和tracrRNA 互補配對的步驟,方便了操作.其他科學家進一步對該系統進行優化,如Gasiunas 等[42]證明,PAM 序列是DNA 結合所必需的,并證明了Cas9D31A和Cas9N891A同樣能產生單鏈切刻.
CRISPR/Cas9 系統的打靶功能一經發現便得到了廣泛應用,2013年是CRISPR 應用的爆發之年.美國博德研究所的張鋒實驗室在人類細胞和小鼠等真核細胞中進行精確基因編輯獲得成功[43],首次實現了CRISPR/Cas9 系統在真核細胞內的應用,對SpCas9和SpRNaseⅢ的編碼序列進行了優化,并添加了核定位信號,使其真核細胞中高效表達,以人類EMX1 位點的30 bp 序列為靶標,通過轉化不同的組合進入人類細胞293FT,實現了對靶標基因EMX1 的編輯.與TALEN 的效率相比,CRISPR/Cas 的效率略高.進一步研究居間序列的點突變對切割效率的影響,發現當點突變發生在PAM 之前的11 個堿基之內時,不會產生切割;當13個堿基處突變時,會有一定效率的切割,表明具有潛在的脫靶可能性.因此提出采用RvuCI 結構域失活的Cas9 可能會降低脫靶.研究者對EMX1位點中相距119 bp 的2 個位點進行打靶,發現有1.6%的機會獲得片段缺失.
在同一期Science 雜志上,哈佛醫學院的Church實驗室也報道了利用CRISPR/Cas9 系統在多個人類細胞系中進行同源重組基因編輯的研究[44].該研究先將一段報告基因序列轉入人類胚腎細胞系HEK293T中,而后設計T1 和T2 兩種gRNA 分別與Cas9 質粒和重組模板一起轉化HEK293T 細胞,得到的編輯效率分別為3%和8%.
隨后,許多實驗室利用CRISPR/Cas9 系統得到了基因編輯的其他模式動物,如基因編輯的斑馬魚[45]、果蠅[46]、非洲蟾蜍(Xenopus tropicalis)[47]等.中國的季維智院士等科學家得到了基因編輯的食蟹獼猴[48].
3.2 CRISPR/Cas9編輯技術在植物研究中的應用
Nature biotechnology 雜志在同一期發表了3 篇CRISPR 在植物中的應用研究.中科院遺傳發育所高彩霞研究組[49]以水稻原生質體為材料,以八氫番茄紅素去飽和酶基因OsPDS 為靶標基因,設計了SP1 和SP2 兩條sgRNA,靶標序列含有PstⅠ酶切位點.根據植物密碼子偏好,優化了Cas9的密碼子,為了保證定位到細胞核中,在其N 端和C 端都加上核定位信號NLS.轉化子經PCR、 酶切、 測序驗證,得到了15%的突變率.之后又對3個水稻基因和一個小麥基因打靶,得到的突變率為26.5%~38.0%.研究者還對CRISPR/Cas9 系統誘導的同源重組效率進行了初步試驗,在29 個轉化子中得到了2 個陽性轉化子,效率為2/29.
美國哈佛醫學院的研究人員[50]以擬南芥原生質體為材料,以擬南芥八氫番茄紅素去飽和酶基因AtPDS3 為靶標基因,比較了植物密碼子優化的Cas9蛋白pcoCas9 和細菌Cas9 蛋白SpCas9的效率,發現前者效率顯著高于后者.對轉化后的擬南芥原生質體PCR測序,得到5.6%的突變率,對另一個基因AtFLS2 打靶,得到的突變率為1.1%.研究還計算了轉化時所用gRNA 和Cas9 蛋白的相對比例,發現當比例為1 ∶1時最有效,當比例為1 ∶19 時無效.只設計一條gRNA對擬南芥AtRACK1b 和AtRACK1c 基因中相同的一段序列進行打靶時,發現2 個基因都發生了突變.在對煙草細胞的NbPDS 基因打靶時,得到了38%左右的突變率.
英國諾維奇科技園的Nekrasov 等[51]用農桿菌侵染煙草葉片的方法在煙草中做了CRISPR/Cas9 系統基因打靶的研究.他們首先將GFP 基因與Cas9 蛋白融合,觀察其核定位效果,而后用擬南芥U6 啟動子驅動的gRNA 與Cas9 基因一起注射煙草,經過PCR、酶切實驗,得到了2.1%的突變率.
此外,還有多位中國科學家將CRISPR 技術應用到植物研究中.如Mao 等[52]研究了靶標位點的PAM 序列對打靶效率的影響,發現不正確的PAM 序列會使效率降低(從11%降到5.4%),但不會消除;Feng 等[53]觀察了用CRISPR/Cas9 系統在擬南芥中打靶后的數個世代,發現T1、T2、T3 代帶有突變的植株分別為71.2%、58.3%和79.4%,突變類型主要是單堿基插入和短的缺失.Miao 等[54]也在水稻中進行了誘導基因突變的研究,獲得了較高的突變率.Ma 等[55]構建了一種可以串聯多個靶點的CRISPR/Cas9 系統,利用Golden gate 方法,最多可以一次將8 個靶點串聯起來進行多基因編輯.
由于CRISPR/Cas 技術操作方便,因此不斷有大量的與該技術相關的論文發表在國際期刊上.后來人們又發現了類似于CRISPR/Cas9 系統的CRISPR/Cpf1系統[56]、具有更高兼容性的PAM 結構和更高DNA 特異性的xCas9 系統[57]等.
4 CRISPR/Cas9技術的優勢及應用要點
4.1 幾種基因編輯方法的比較
CRSPR/Cas 系統原理簡單,設計方便,已在多種動物和植物中得到了應用,它與ZFN 和TALEN 的比較結果如表1 所示.CRSPR/Cas 系統具有以下優勢:①設計更為方便.ZFN 和TALEN 需要考慮DNA 雙鏈的結構特征,還要考慮2 個位點之間spacer 的距離,CRISPR/Cas 系統則只需一條鏈上帶有PAM 結構.②構建更為便捷.TALEN 需要多個串聯重復單體的組裝,CRISPR/Cas 系統只需構建長度約為100 堿基的gRNA.③CRISPR/Cas 系統可多次作用,一旦將Cas蛋白整合到染色體上,再次利用時,只需重新導入短片段的gRNA.④可對多個基因打靶.由于gRNA 序列很短,容易構建,因而一次可將多個基因的gRNA 轉入植物中實現對多個基因的編輯.
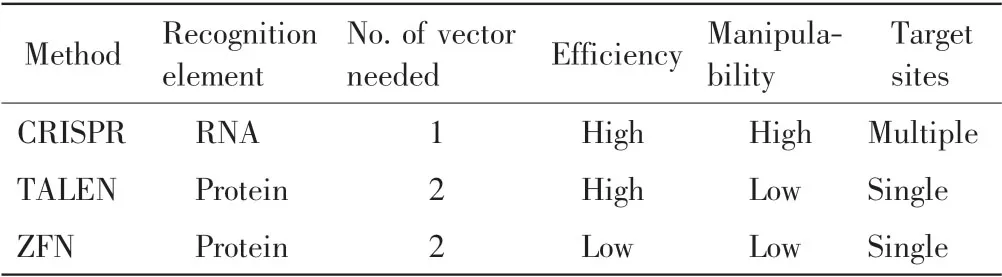
表1 3 種基因編輯技術的比較Tab.1 Comparison of three gene editing techniques
4.2 CRISPR/Cas9的應用要點
CRISPR/Cas9 已成為分子生物學研究的必備工具.在理解其作用原理的基礎上,具體運用中把握住以下要點,才能更高效地發揮作用,達到事半功倍的效果.
(1)Cas9 蛋白的密碼子優化.不同生物具有密碼子偏好性,CRISPR/Cas 系統來源于古細菌,應用時要根據目標物種的偏好性進行優化,這樣轉進去的Cas9才能夠得到高效的翻譯.Li 等[50]以擬南芥原生質體為材料,以擬南芥八氫番茄紅素去飽和酶基因AtPDS3為靶標基因,比較了植物密碼子優化的pcoCas9 蛋白和細菌的SpCas9 蛋白的效率,發現前者的效率顯著高于后者.
(2)啟動子選擇.選擇適合于目的生物的啟動子,保證其能高效驅動Cas9 蛋白和gRNA 的轉錄.目前常用的啟動子是泛素連接酶的Ubi 啟動子,人類細胞系中常用的是U6 啟動子[44],植物中常用的是U6 或U3啟動子[49].
(3) 靶位點的選擇.gRNA 的選擇一般為20 個堿基,其3’ 端之后一般是NGG 的結構.在人類細胞系中一般為GN20NGG(U6 啟動子)[44];在植物中一般為5’-A-N(20)GG-3’(U3 啟動子)或5’-G-N(20)GG-3’(U6 啟動子)[49].對于真核生物來說,靶位點應該位于其外顯子序列上.研究者開發了數個用于gRNA 在線設計的平臺,其中大多數平臺既可以用來設計靶位點,還能給出潛在的脫靶位點,如https://omictools.com/casot-tool、http://www.e-crisp.org/E-CRISP/、http://skl.scau.edu.cn/等.如果目的是敲除基因的功能,靶位點最好選擇其5’上游,這樣可以產生移碼突變或者提前終止;如果編碼基因很長,可以把靶位點選擇在保守結構域上.靶位點的選擇除了需滿足上述要求,還應該考慮編輯后便于檢測.切割位點一般位于NGG前1~4 個堿基處,可以選擇在此處有酶切位點的靶序列.編輯后酶切位點被破壞,可通過PCR 擴增之后用酶切電泳來篩選結果[49].
(4)編輯結果分析.轉化后,可提取被轉化材料的DNA,在靶位點兩側設計引物,進行PCR 擴增初篩.如果靶序列中有酶切位點,可以利用酶切產物電泳來進行初步篩選,之后進行測序;對于沒有酶切位點的靶序列,將擴增產物與野生型的擴增產物混合后退火,則會形成不完全互補的DNA 雙鏈,再用單鏈核酸內切酶SⅠ酶切,對經過初篩后的陽性克隆進行測序分析.
基因編輯結果大致有3 種情況,純合基因型、雜合基因型和雙等位突變.純合基因型最容易分析,與野生型比對即可看出是插入或者缺失.對于雜合型和雙等位突變,可以通過挑選單克隆測序進行分析,也可以對測序結果峰圖進行分析.劉耀光研究組開發的平臺同樣可以用于對測序結果峰圖的分析(http://skl.scau.edu.cn/dsdecode/).
5 結語
基因編輯技術的發展為基因治療和生物性狀改良帶來了新的機遇.人們有望通過基因編輯技術,對患者來源的誘導多能干細胞(Induced pluripotent stem cells,iPSCs) 進行編輯,進而治療包括杜氏肌營養不良癥、帕金森疾病等在內的多種遺傳疾病以及艾滋病、乙型肝炎等傳染性疾病,但仍有脫靶效應、新的基因導入系統等技術瓶頸[58-59].最近,Liang 等[60]發明了一種將CRISPR/Cas mRNA 包裹為體外轉錄產物(in vitro transcripts,IVTs)或核糖蛋白復合體(Ribonucleoprotein complexes,RNPs) 后再轉入植物幼胚的方法,獲得了外源DNA-free 的小麥基因敲除植株.由于該方法沒有外源DNA 進入植物體,與以往的基因改良生物有很大區別,因而可能在食品安全立法時予以豁免.總之,基因編輯技術已經獲得了巨大進展,但利用其改變人類的生命和生活仍有許多難題需要克服[61].
猜你喜歡
工業設計(2022年8期)2022-09-09 07:43:20
軍民兩用技術與產品(2021年10期)2021-03-16 06:05:30
北京測繪(2020年12期)2020-12-29 01:33:58
裝備制造技術(2019年12期)2019-12-25 03:06:46
制造技術與機床(2019年10期)2019-10-26 02:47:06
中國洗滌用品工業(2019年4期)2019-05-11 09:27:34
鐵道通信信號(2018年5期)2018-06-28 03:06:24
家庭影院技術(2017年9期)2017-09-26 03:41:45
知識經濟·中國直銷(2017年5期)2017-06-15 20:28:19
通信電源技術(2016年6期)2016-04-20 06:21:32
