含添加劑的二步溶液法制備鈣鈦礦太陽能電池
2019-06-14 08:02:08鄭海松魏愛香肖志明
發光學報 2019年6期
關鍵詞:效率
鄭海松,魏愛香,劉 俊,肖志明,招 瑜
(廣東工業大學材料與能源學院,廣東廣州 510006)
1 引 言
有機/無機鹵化物鈣鈦礦太陽能電池(PSCs)由于其光電轉換效率(PCE)在短短幾年內從3.8%[1]迅速提高至 22.1%[2]而備受關注。然而,傳統結構的PSCs也面臨著一些問題,例如真空蒸發技術制備金電極和昂貴的小分子材料spiro-OMeTAD作為空穴傳輸層均會導致電池的制備成本過高,阻礙了未來PSCs的商業化應用。因此,降低成本和提高穩定性是當務之急。一方面,碳材料來源廣泛且價格便宜,如石墨和石墨烯具有良好的導電性且易于制備;另一方面,鈣鈦礦材料本身既可以被用作光吸收層,也可以作為空穴傳輸層[3-8],因此,基于碳電極的無空穴傳輸層的鈣鈦礦太陽能電池應運而生。
在一系列用于制備高質量鈣鈦礦吸收層的方法中,二步溶液法簡單且成本低,二步溶液法不僅比一步溶液法更容易控制鈣鈦礦的結晶過程,而且與真空蒸鍍和氣相輔助溶液工藝制備方法相比,不需要用到昂貴的實驗設備,可大幅度降低成本[9-15]。然而,用傳統的碘化鉛/N,N-二甲基甲酰胺(PbI2/DMF)溶液制備的PbI2薄膜浸泡在甲基碘化胺/異丙醇(MAI/IPA)溶液中,通常需要較長的時間才能把PbI2完全轉變為鈣鈦礦(CH3NH3PbI3)[16],這會使鈣鈦礦吸收層表面形成較大的鈣鈦礦晶粒[17],導致鈣鈦礦吸收層與碳電極之間接觸不良。同時,一些殘留在鈣鈦礦薄膜中的PbI2會降低鈣鈦礦太陽能電池的穩定性[18]。
本文使用含二甲基亞砜(DMSO)添加劑的二步溶液法制備高質量CH3NH3PbI3吸收層,并制備結構為 FTO/TiO2致密層/TiO2介孔層/CH3NH3PbI3吸收層/碳電極的鈣鈦礦太陽能電池。研究浸泡時間和浸泡濃度對CH3NH3PbI3薄膜以及PSCs的光伏性能的影響規律。我們先前的研究工作已表明[19]:采用濃度為1.0 mol/L的PbI2/DMF溶液制備的PbI2薄膜在0.063 mol/L的MAI/IPA溶液中需浸泡3.5 h后,PbI2才能完全轉化為 CH3NH3PbI3,延長浸泡時間會在CH3NH3PbI3表面形成個別較大的晶粒,導致鈣鈦礦/碳電極界面接觸不良,從而導致PSCs的填充因子較低。為了獲得高質量的鈣鈦礦層,提高PSCs的填充因子和光電轉換效率,本文使用DMSO和DMF的混合溶劑代替純DMF溶劑配制Pb I2前驅溶液用于制備PbI2薄膜,由于加入添加劑DMSO之后,使PbI2薄膜呈多孔疏松狀態,從而使PbI2完全轉化為CH3NH3PbI3的時間縮短為30 min,且改善了CH3NH3PbI3/碳電極的接觸,提高了PSCs的填充因子和轉換效率。
2 實 驗
2.1 TiO2致密層和介孔層的制備
首先把摻F的SnO2透明導電玻璃(FTO)依次放入丙酮和無水乙醇中超聲清洗25 min,然后用氮氣槍吹干。將1 g的75%雙(乙酰丙酮基)二異丙基鈦酸酯與10.3 g正丁醇充分攪拌混合,形成TiO2致密層前驅液。在500 r/min、3 s,3 000 r/min、30 s的條件下將前驅液旋涂在預熱好的FTO上,然后100℃加熱15 min,再經過500℃退火形成TiO2致密層。以18NR-T與無水乙醇質量比為1∶4的比例稀釋18NR-T,形成TiO2介孔層旋涂漿料。在5 000 r/min、30 s的條件下將漿料旋涂在預熱好的致密層上,然后100℃加熱15 min,最后經過500℃退火15 min形成TiO2介孔層。
2.2 鈣鈦礦吸收層的制備
將461 mg的PbI2粉末溶于200μL DMSO與800μLDMF混合溶劑中,攪拌5min,然后放到保濕柜里靜置2.0 h,待PbI2粉末完全溶解即可形成濃度為1.0 mol/L的Pb I2溶液。在500 r/min、3 s,3 500 r/min、30 s 的條件下將 PbI2溶液旋涂在預熱好的介孔層上,然后100℃加熱40 min后得到PbI2薄膜。為了研究不同浸泡時間和不同浸泡濃度對鈣鈦礦層的形貌和結構以及PSCs的光伏性能的影響規律,設計了兩組實驗。
A組:將100 mg的MAI粉末溶于10 mL的IPA中,攪拌30 min,待MAI粉末完全溶解即可形成0.063 mol/L的MAI/IPA溶液。將PbI2薄膜置于 MAI/IPA 溶液中分別浸泡 8,12,20,30,40,60,90min。然后用異丙醇清洗樣品表面,去除多余的MAI。最后100℃加熱40 min,得到CH3NH3PbI3吸收層。
B 組:分別將 70,100,130 mg的 MAI粉末溶于10 mL的IPA中,攪拌30 min,待MAI粉末完全溶解即可形成濃度分別為 0.044,0.063,0.082 mol/L的MAI/IPA溶液。將PbI2薄膜分別置于不同濃度的MAI/IPA溶液中浸泡40 min。然后用異丙醇清洗樣品表面,去除多余的MAI。最后100℃加熱40 min,得到CH3NH3PbI3吸收層。
采用場發射掃描電子顯微鏡(FE-SEM,SU8010)和X射線衍射儀(XRD,MAX-Ultima IV)對 TiO2致密層、TiO2介孔層、PbI2薄膜和CH3NH3PbI3吸收層的形貌和結構進行表征。XRD的測試條件為:CuKα射線,λ=0.154 2 nm,掃描范圍 10°~60°,掃描速度為6(°)/min。
2.3 鈣鈦礦太陽能電池的制備和光伏性能測試
第一步,在清洗干凈的FTO上旋涂制備TiO2致密層;第二步,在致密層上旋涂制備TiO2介孔層;第三步,在介孔層上制備Pb I2薄膜;第四步,將PbI2薄膜浸泡到MAI/IPA溶液中生成CH3NH3PbI3層;第五步,在吸收層上刮涂一層碳漿料,在100℃干燥30 min后得到結構為FTO/TiO2致密層/TiO2介孔層/CH3NH3PbI3吸收層/碳電極的鈣鈦礦太陽能電池。上述實驗過程全部在空氣中進行。采用Keithley 2400數字源表測試電池的J-V特性曲線,所用的光源為500 W的氙燈(AM-1.5),光功率密度調整和校準為100mW/cm2,電池的有效受光面積為0.16 cm2;并采用太陽能電池外量子效率測試儀進行光譜響應特性分析;采用電化學工作站進行電化學阻抗譜分析;采用紫外可見分光光度計進行吸光度和透過率分析。
3 結果與討論
3.1 浸泡時間對鈣鈦礦層結構和形貌的影響
圖1是在不同浸泡時間下制備的CH3NH3PbI3吸收層的XRD衍射圖。由圖1可知,當PbI2薄膜在濃度為0.063mol/L的MAI/IPA溶液中的浸泡時間小于30 min時,雖然大部分PbI2已經轉化為 CH3NH3PbI3,但是在2θ=12.67°處出現微弱的衍射峰,該峰對應于PbI2的(001)晶面衍射峰(JCPDSNo.07-0235),說明仍有少量 PbI2殘留。位于 14.04°、19.94°、23.46°、24.48°、28.32°、31.86°、34.92°、40.52°和 43.04°處的 XRD 衍射峰分別對應 CH3NH3PbI3的(110)、(112)、(211)、(202)、(220)、(310)、(312)、(224)和(314)晶面的衍射峰[20]。浸泡時間為30 min時,PbI2的衍射峰消失,表明PbI2已經完全轉化為CH3NH3PbI3。當浸泡時間為40 min時,鈣鈦礦的特征峰最強,說明此時鈣鈦礦的結晶性最好。結果表明浸泡時間會影響CH3NH3PbI3吸收層的相純度。
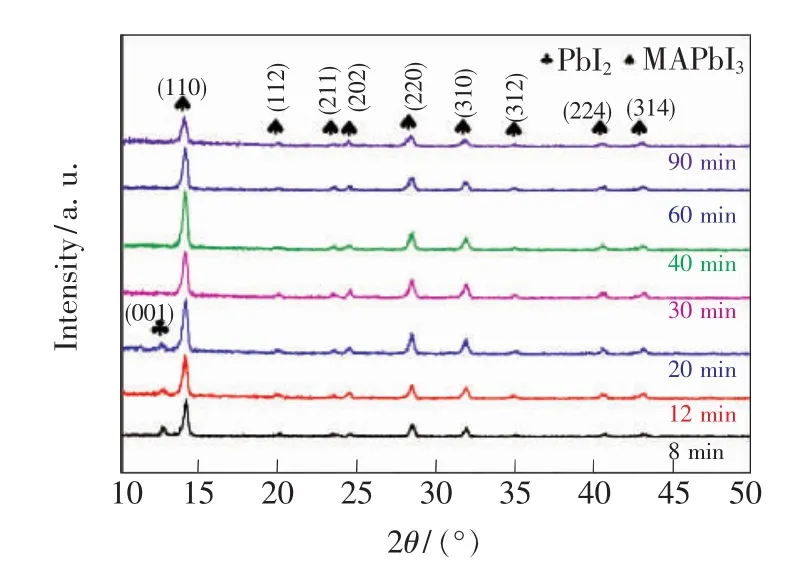
圖1 不同浸泡時間制備的CH3 NH3 PbI3吸收層的XRD衍射圖Fig.1 X-ray diffraction pattern of the CH3 NH3 PbI3 layerprepared at different dipping times
圖2 (a)和(b)分別是采用純DMF溶劑和DMSO/DMF混合溶劑制備的PbI2薄膜的SEM圖像,可以看出加入DMSO之后PbI2薄膜孔隙率增加,表面更加疏松,有利于MAI/IPA溶液滲入PbI2薄膜內部,縮短 PbI2完全轉換成CH3NH3PbI3的時間。Wu等[21]研究了 Pb I2與溶劑分子 DMF和 DMSO之間的配位關系,發現DMF-PbI2形成絡合物時,Pb—O鍵長為0.243 1 nm,Pb2+與DMF溶劑的配位比為1∶1;DMSOPbI2形成絡合物時,Pb—O鍵長為0.238 6 nm,Pb2+與DMSO溶劑的配位比為1∶2。表明DMSO與PbI2的配位能力比DMF與PbI2的配位能力強。因此,采用PbI2/DMF溶液制備 PbI2薄膜時,由于DMF-PbI2絡合物中Pb—O鍵比較弱,旋涂過程中溶劑容易揮發,導致PbI2在退火之前就會在TiO2介孔層上結晶。DMSO-Pb I2絡合物的沸點較高以及DMSO和Pb2+之間較強的相互作用,能減緩溶劑在旋涂過程中的揮發。高溫下退火時,殘余的DMSO溶劑分子將逐漸揮發,溶劑分子在退火過程進一步釋放,導致PbI2薄膜呈多孔疏松結構,如圖2(b)所示。圖2(c)~(i)分別為 PbI2薄膜在濃度為0.063 mol/L的MAI/IPA 溶液中浸泡 8,12,20,30,40,60,90 min得到的CH3NH3PbI3薄膜的SEM圖像。當浸泡時間為8~40 min時,鈣鈦礦薄膜的表面呈現立方體形狀的晶粒,鈣鈦礦層由尺寸均勻的晶粒組成,表面光滑。然而,當浸泡時間為60 min和90 min時,在鈣鈦礦層的表面上會形成一些比較大的鈣鈦礦晶粒,從而出現粗糙的鈣鈦礦層。

圖2 (a,b)采用純DMF溶劑和DMSO與DMF混合溶劑制備的PbI2薄膜的SEM像;(c~i)不同浸泡時間得到的CH3 NH3 PbI3的SEM像。Fig.2 (a,b)SEM images of PbI2 films prepared using pure solventof DMF andmixed solventof DMFwith DMSO,respectively.(c-i)SEM images of CH3 NH3 PbI3 layer prepared at different dipping times.
3.2 浸泡濃度對鈣鈦礦層結構和形貌的影響
圖3 是將PbI2薄膜分別浸入濃度為0.044,0.063,0.082 mol/L 的 MAI/IPA 溶液中浸泡 40 min得到的CH3NH3PbI3吸收層的XRD衍射圖。隨著MAI/IPA溶液濃度的增加,PbI2更快地轉化為CH3NH3PbI3。由圖3可看出,PbI2薄膜在濃度為0.044 mol/L的 MAI/IPA溶液中浸泡40 min后,仍有PbI2殘留在鈣鈦礦層中。而PbI2薄膜在0.063 mol/L和0.082 mol/L的 MAI/IPA 溶液里浸泡40 min后均可完全轉化為鈣鈦礦。結果表明,在增大MAI/IPA溶液的濃度后,PbI2轉化為CH3NH3PbI3的速度明顯加快。
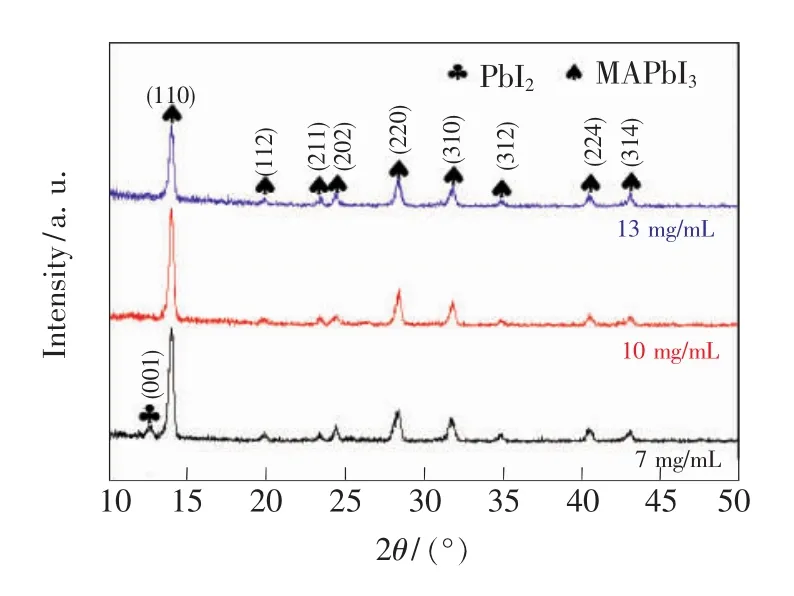
圖3 PbI2薄膜在不同濃度的MAI/IPA溶液中浸泡40 min得到的CH3 NH3 PbI3的XRD圖Fig.3 X-ray diffraction pattern of the CH3 NH3 PbI3 prepared by dipping PbI2 films in MAI/IPA solution with different concentrations

圖4 PbI2薄膜在不同濃度的MAI/IPA溶液中浸泡40 min得到的CH3 NH3 PbI3的SEM圖像Fig.4 SEM images of CH3NH3 PbI3 layer prepared by dipping PbI2 films in MAI/IPA solution with different concentration
圖4(a)、(b)、(c)是通過將PbI2薄膜分別浸入0.044,0.063,0.082 mol/L 的 MAI/IPA 溶液中浸泡40min得到的CH3NH3PbI3的SEM圖像。在0.044,0.063mol/L的MAI/IPA溶液中浸泡40min得到的鈣鈦礦薄膜表面非常平整,由尺寸均勻的晶粒組成。但PbI2薄膜在濃度為0.082mol/L的MAI/IPA溶液中浸泡40min后生成的鈣鈦礦層表面則出現了少數大晶粒。結合圖3和圖4可知,在濃度為0.082mol/L的MAI/IPA溶液里浸泡40 min后已經屬于過度浸泡,其結晶性反而降低。
3.3 鈣鈦礦太陽能電池的光伏性能及阻抗譜分析
在TiO2致密層、介孔層和碳電極制備工藝相同的條件下,以PbI2薄膜在濃度0.063 mol/L的MAI/IPA 溶液中分別浸泡20,30,40,60,90 min 后得到的鈣鈦礦層作為吸收層,制備鈣鈦礦太陽能電池,這5個電池編號分別為 SC1A、SC2A、SC3A、SC4A和SC5A。以PbI2薄膜分別浸泡在濃度為0.044,0.063,0.082mol/L 的 MAI/IPA 溶液中40 min 得到的CH3NH3PbI3層作為吸收層,制備鈣鈦礦太陽能電池,這3個電池編號分別為SC1B、SC2B和SC3B。圖5(a)、(b)為兩組電池的J-V特性曲線。圖5(c)為電池SC2B的外量子效率特性曲線(IPCE)。圖5(d)為 A組電池的電化學阻抗譜(EIS)。圖5(e)為電池SC1B和SC2B的紫外可見吸收和透射光譜。圖5(f)為電池SC2B的截面SEM圖,由此可得到TiO2致密層厚度約為70 nm,介孔層厚度約為500 nm,鈣鈦礦層厚度約為550 nm。
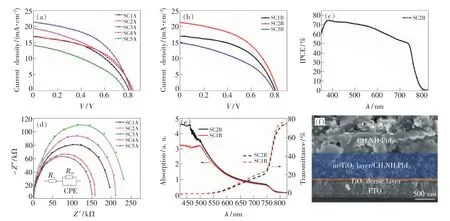
圖5 (a,b)鈣鈦礦太陽能電池的J-V特性曲線;(c)電池SC2B的外量子效率特性曲線;(d)鈣鈦礦電池的電化學阻抗譜;(e)電池SC1B和SC2B的紫外可見吸收和透射光譜;(f)電池SC2B的截面SEM圖。Fig.5 (a,b)J-V curves of PSCs.(c)IPCE spectra of SC2B.(d)EISspectra of PSCs.(e)UV-vis absorption and transmittance spectra of the SC1B and SC2B.(f)Cross-sectional SEM image of SC2B.
表1為根據圖5(a)、(b)得到的兩組鈣鈦礦太陽能電池的光伏特性參數。從圖5(a)和表1可看出,隨著PbI2薄膜在MAI/IPA溶液中浸泡反應時間的延長,PbI2含量相應減少,鈣鈦礦層的結晶性逐漸增強,電池的光伏性能逐漸提高。A組電池中,當浸泡時間為40 min時制備的電池(SC3A)達到了最佳的光伏性能,即開路電壓(Voc)達到0.82 V,短路電流密度(Jsc)達到 21.21 mA/cm2,填充因子(FF)達到0.49,光電轉化效率(PCE)提高至8.61%,且在整個可見光區的光子-電子的轉換效率接近70%。但是當繼續延長浸泡時間到60 min以上時,電池的光伏性能反而有所下降,主要原因是過度浸泡后鈣鈦礦層表面出現比較大的鈣鈦礦晶粒,使得表面平整性下降,導致鈣鈦礦層與碳電極的接觸較差,從而使填充因子和短路電流密度減小,最終導致光電轉換效率降低。從圖5(d)可看出,A組電池中,SC3A具有最低的串聯電阻Rs和電荷傳輸阻抗Rct,與其具有最大的填充因子和短路電流密度的實驗結果相一致[22]。從圖5(b)和表1可看出,B組電池中,在相同的浸泡時間下,電池SC1B由于仍有一部分PbI2殘留在鈣鈦礦層中,阻礙載流子的遷移,導致其短路電流密度較低;此外,從圖5(e)可看出,SC2B的鈣鈦礦層在可見光范圍內對光的吸收能力比SC1B強。SC3B由于過度浸泡,鈣鈦礦層與碳電極之間的接觸不良,二者之間較大的串聯電阻導致短路電流密度和填充因子減小。所以電池SC1B和SC3B的光電轉化效率均沒有電池SC2B的轉換效率高。
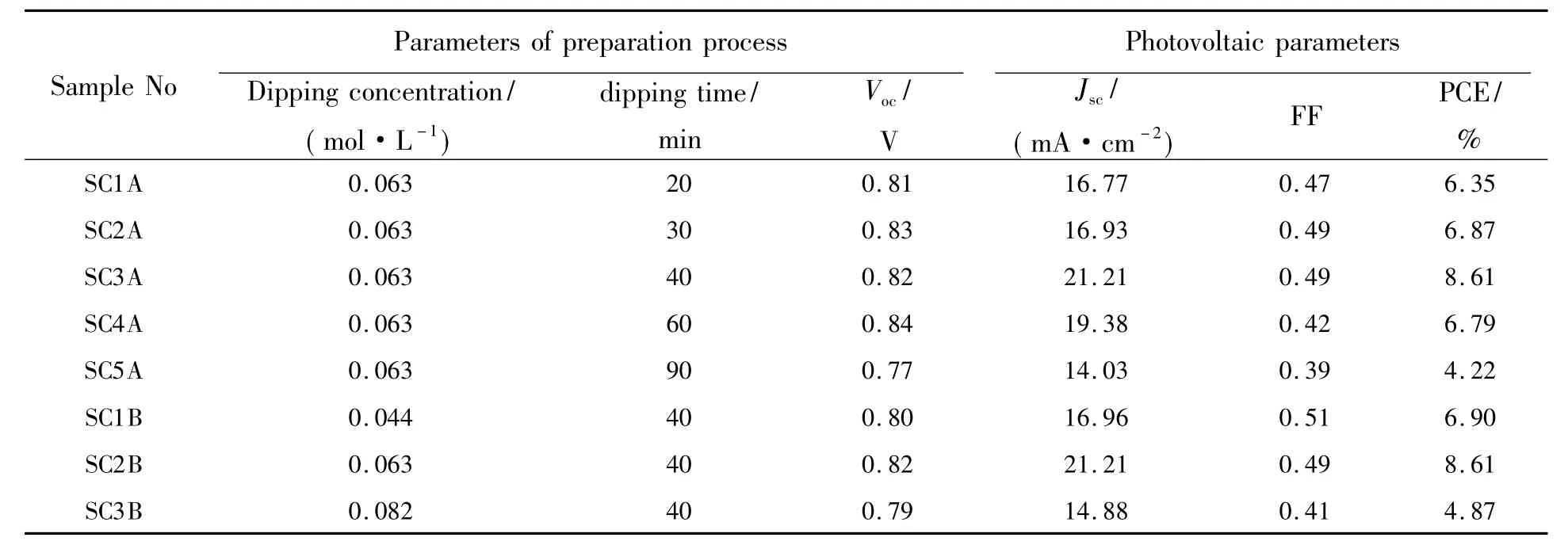
表1 鈣鈦礦太陽能電池的制備工藝條件和光伏特性參數Tab.1 Parameters of preparation process and photovoltaic characteristics of PSCs
目前基于碳電極的無空穴傳輸層的鈣鈦礦太陽能電池的最高效率已達到14.5%[23]。從表1可知,本文制備的電池與目前最高效率的相同結構的電池相比,主要是填充因子較低,導致光電轉換效率較低。填充因子低的主要原因是鈣鈦礦/碳電極界面的接觸差和串聯電阻較大,提高電池的填充因子和光電轉換效率主要從以下兩個方面進行改進:(1)制備覆蓋率高、致密性好且表面光滑的鈣鈦礦吸收層;(2)改進碳電極的制備工藝,例如采用導電性更好的石墨烯制備漿料,或將石墨烯直接制備或壓制在鈣鈦礦吸收層上,改善鈣鈦礦/碳電極的接觸,降低串聯電阻,從而提高電池的效率。
4 結 論
本文以PbI2粉末溶于DMSO與DMF混合溶劑中形成的前驅液制備PbI2薄膜,然后將Pb I2薄膜在相同濃度的MAI/IPA溶液中浸泡不同時間、以及在不同濃度的MAI/IPA溶液中浸泡相同時間后得到的CH3NH3PbI3薄膜作為吸收層,制備基于碳電極的無空穴傳輸層的鈣鈦礦太陽能電池。研究了浸泡時間和MAI/IPA溶液的濃度對PSCs光伏性能的影響。結果表明,適當提高PbI2薄膜在MAI/IPA溶液中的浸泡時間,有利于提高鈣鈦礦層的相純度和結晶性,進而提高電池的光伏性能。當浸泡時間為40 min時,鈣鈦礦太陽能電池獲得了最佳的光伏性能,其開路電壓為0.82 V,短路電流密度為21.21 mA/cm2,填充因子為0.49,光電轉化效率為8.61%,且在整個可見光區的光子-電子的轉換效率接近70%。但是當浸泡時間過長,CH3NH3PbI3薄膜表面會出現大晶粒,導致鈣鈦礦層和碳電極之間接觸變差,使得填充因子降低,從而導致電池光電效率降低。而在相同的浸泡時間下,MAI/IPA溶液的濃度則會顯著影響PbI2轉化成CH3NH3PbI3的速度,MAI/IPA溶液的濃度越高,PbI2完全轉化成CH3NH3PbI3的速度越快。
猜你喜歡
瘋狂英語·初中天地(2021年5期)2021-07-21 02:24:28
甘肅教育(2020年14期)2020-09-11 07:57:42
中學生數理化(高中版.高考數學)(2020年5期)2020-06-02 09:19:08
商周刊(2017年9期)2017-08-22 02:57:49
遼寧經濟(2017年6期)2017-07-12 09:27:16
中國衛生(2016年9期)2016-11-12 13:27:54
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國洗滌用品工業(2015年7期)2015-02-28 19:02:38
電子設計工程(2015年12期)2015-02-27 12:06:10
中國衛生(2014年11期)2014-11-12 13:11:32
