利用染色質開放性測序技術探討葉酸缺乏對胚胎干細胞基因組結構變化調控影響
2019-06-24 08:42:40謝秋李才華王滔孫正怡郁琦張霆
生殖醫學雜志 2019年6期
謝秋,李才華,王滔,孫正怡,郁琦,張霆
(1.中國醫學科學院 北京協和醫學院 北京協和醫院,北京 100730;2.上海天昊生物,上海 200120;3. 首都兒科研究所,北京 100020)
真核生物染色體以核小體為單位進行致密折疊,同時部分染色質會呈現開放狀態,這些開放結構通常作為特異性反式作用因子(如轉錄因子、酶等)和順式作用元件(如增強子、隔離子等)與基因組DNA相關作用的活躍區域。染色體的開放程度用染色體可接近性(Chromatin Accessibility)來表示,該狀態的變化會對基因表達調控、DNA復制和修復等產生重要影響。之前有研究報道的染色質開放性測序技術(Assay for Transposase Accessible Chromatin with high throughput sequencing,ATAC-seq)方法,利用高靈敏度的轉座酶(Transposase,Tn5)在尋找染色質可接近位置的同時對染色體DNA進行片段化,細胞數量級在10的四次方即可[1-2]。因此對于數量有限的臨床樣本尤其適合,實驗操作上更方便快捷,是一種創新的表觀遺傳學研究技術手段。流行病學研究表明圍孕期葉酸缺乏是習慣性流產、出生缺陷發生的重要危險因素,補充葉酸能有效預防上述不良反應的發生[3-5]。越來越多的研究表明葉酸作為一碳單位的運載體作為甲基供體影響表觀修飾調控[6-8]。本研究通過ATAC-seq技術研究葉酸代謝異常對小鼠胚胎干細胞(mouse embryonic stem cell,mESC)全基因組染色質開放程度的影響,為探索葉酸缺乏對早期胚胎發育表觀編程影響提供初步的研究基礎。
資料與方法
一、研究對象及試劑
1.研究對象:Sv/129 mESC細胞來源于北京宣武醫院,采用高糖DMEM培養,含15% 胎牛血清、β-巰基乙醇、非必須氨基酸、谷氨酸及白血病抑制因子,培養在預先包被有2% 明膠的培養瓶或者培養板中,37℃、5%CO2培養箱傳代培養,每隔2~3 d傳代1次。
2.主要試劑:甲氨蝶呤(MTX,CalBiochem,美國);Tn5轉座酶(北京MDTKBio);高保真PCR擴增試劑盒(Next High Fidelity 2×PCR Master Mix,NEB,美國);DNA文庫制備試劑盒(Nextera DNA Sample Prep Kit,Illumina,美國);雙末端簇生成試劑盒(v3-cBot-HS,Illumina,美國);核酸純化試劑盒(AgencourtAMPure XP,Beckman Coulter,美國);核酸片段篩選試劑盒(AgencourtSPRIselect Reagent Kit,Beckman Coulter,美國)。
3.主要儀器:PCR儀(ABI2720,美國);低溫離心機(Eppendorf 5810R,美國);核酸蛋白定量儀Qubit3.0(Thermo,美國);磁力架DynaMag(Thermo,美國);倒置顯微鏡ECLIPSETi(Nikon,日本);二代測序儀器Illumina Hiseq 2500(Illumina,美國)。
二、實驗方法
1.細胞計數與裂解:分別使用葉酸濃度為4 mg/ml的正常葉酸培養基和終濃度為0.12 μM的MTX處理mESC 24 h(分別為正常對照組和MTX實驗組),收集細胞。制備PBS重懸的單細胞懸液,0.4%臺盼藍染色后倒置相差顯微鏡下計數,取104個細胞至新的離心管;4℃低溫500g離心5 min,小心棄去上清,用預冷的PBS洗滌1次;離心后重懸于50 μl預冷的Lysis buffer,輕柔吹打分散細胞;棄去上清,置于冰上準備轉座反應。
2.轉座反應與純化:配制如下反應體系,吹打混勻后重懸上一步細胞核沉淀:5×reaction buffer 10 μl,Tn5 Enzyme 5 μl,ddH2O 35 μl。反應條件:37℃孵育30 min。使用純化試劑盒純化上一步產物,操作按試劑盒說明書進行;DNA洗脫液洗脫得到11 μl純化產物。
3.PCR放大與純化:對轉座文庫進行放大和添加樣本特異的Index,配制如下反應體系:上一步轉座DNA 11 μl,10 M Primer F 2 μl,10 M Barcoded Primer R 2 μl,NEBNextUltraTMII Q5Master Mi 15 μl。反應條件:72℃延伸5 min;98℃變性30 s(1個cycle);98℃變性10 s;63℃退火30 s;72℃延伸1 min(10個cycle);72℃延伸5 min;4℃冷卻。使用純化試劑盒純化上一步產物,操作按試劑盒說明書進行;得到最終上機文庫。
4.文庫質控與上機:取1 μl文庫使用Qubit3.0檢測文庫濃度;將不同文庫按照等摩爾量混樣按DNA文庫制備試劑盒操作后上機至Illumina Hiseq平臺,采用PE150模式測序。引物與接頭信息見表1。

表1 文庫構建引物與接頭信息
*IIIIIIII為8 bp的Barcode信息
三、數據分析及統計學處理
測序得到的原始測序序列(raw reads或raw data),里面含有帶接頭的、低質量的reads,為了保證信息分析質量,必須對raw reads進行過濾,得到clean reads用于后續分析。利用trim_galore(http:∥www.bioinformatics.babraham.ac.uk/projects/trim_galore/)進行質量過濾[1,9],包括3個步驟:(1)去掉測序引物;(2)去掉末端低質量的reads;(3)去掉上述1和2步驟后片段<35 bp的reads。正常對照組和MTX實驗組分別設置3個生物學重復,參照小鼠基因組mm10數據庫進行生物信息學分析。
數值分析采用組間均數比較的單因素方差分析,組間計量資料的比較采用獨立樣本t檢驗,P<0.05表示差異有統計學意義。
結 果
一、Tn5轉座酶消化mESC細胞
為了摸索適合的Tn5酶體系,我們選擇兩個酶濃度梯度,分別為5 U和7.5 U,具體標記見表2。
經歷12個cycle擴增后,取少量產物進行瓊脂糖電泳發現Tn5濃度為7.5 U,正常對照組轉座酶消化后核小體周期條帶明顯優于5 U處理濃度,而對于MTX實驗組轉座酶5 U處理優于7.5 U處理,文庫可見清晰的核小體周期條帶(圖1箭頭所示)。

表2 Tn5酶體系摸索
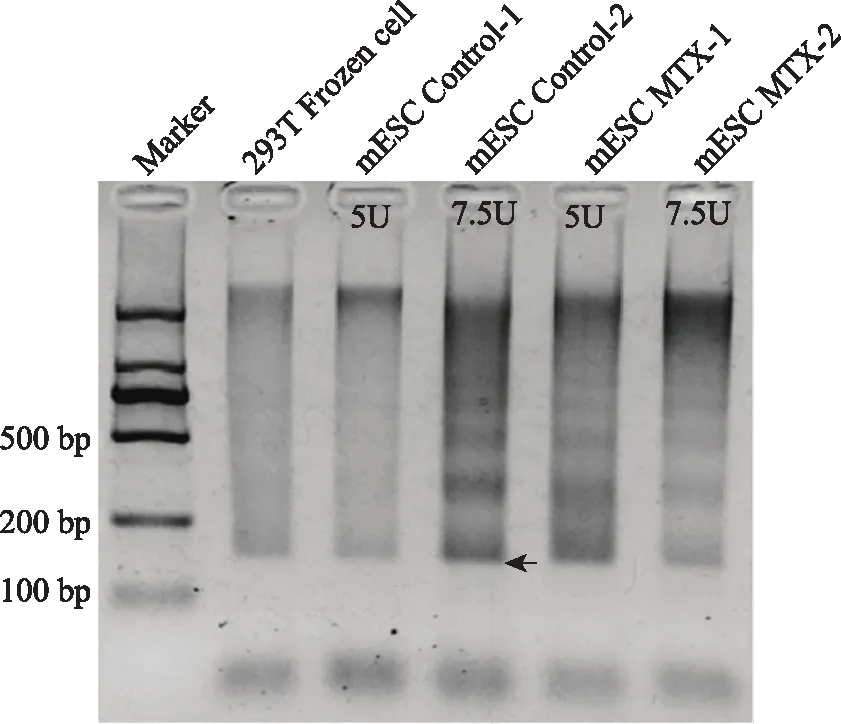
采用5 U、7.5 U Tn5轉座酶分別處理正常對照組、MTX實驗組后檢測核小體分布圖1 Tn5轉座酶消化產物經瓊脂糖凝膠電泳質控
二、有效peak calling總數分析
使用MACS(http:∥liulab.dfci.harvard.edu/MACS/)軟件[10]識別reads在參考基因組上的富集區域,即peak calling,作為候選的蛋白質結合位點(或表觀修飾位點)。正常對照組peak calling數量明顯多于MTX實驗組,中位長度為302 bp(圖2A);MTX實驗組peak calling中位長度為271 bp(圖2B),兩組peak calling數量采用單因素方差分析具有統計學差異(P<0.01),提示MTX處理后mESC染色質可接近區域發生減少。
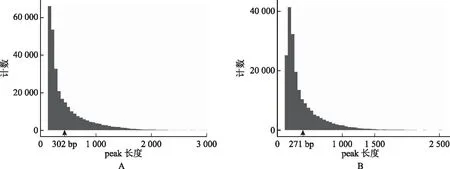
A:正常對照組peak calling計數及長度分布,黑色箭頭示中位peak長度(bp);B:MTX實驗組peak calling計數及長度分布,黑色箭頭示中位peak長度(bp)圖2 兩組peak calling數量及長度分布示意圖
三、差異peak calling基因本體論富集分析
基因本體論(Gene Ontology,GO)是基因功能國際標準分類體系,對目標區域基因進行篩選與分類富集。對正常對照組和MTX實驗組差異peak calling進行GO富集分析比較,發現MTX處理后差異染色質開放區域主要集中胚胎器官形成、細胞周期調控、神經骨骼系統發育及生殖系統發育有關基因(表3)。
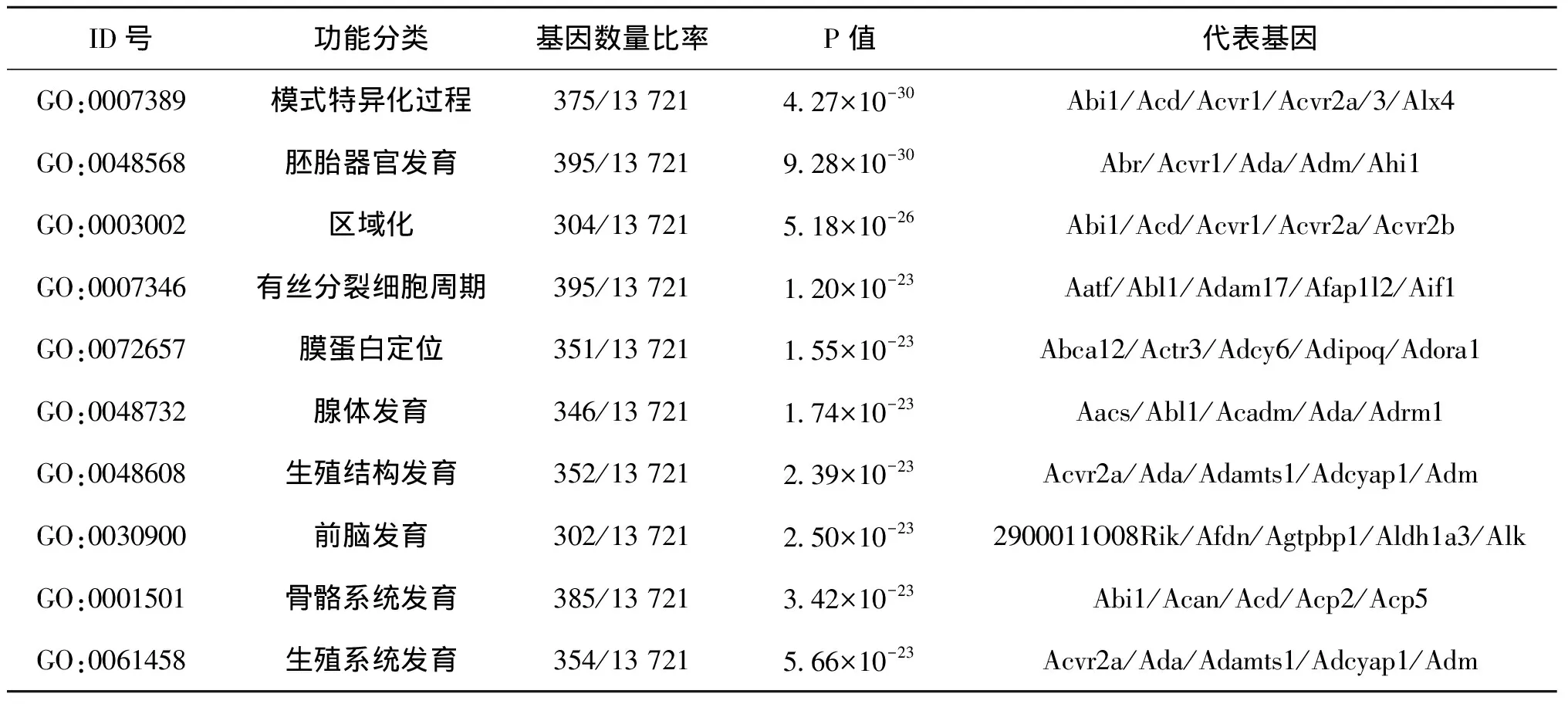
表3 差異peak相關區域GO富集顯著性差異分析(Top 10)
四、差異peak相關基因KEGG富集分析
我們對差異peak相關基因進一步進行KEGG(Kyoto Encyclopedia of Genes and Genomes) Pathway分析,通過Pathway顯著性富集分析能確定差異peak相關基因集合主要參與的生化代謝通路和信號轉導通路。正常對照組和MTX實驗組差異peak 相關基因顯著性富集的pathway集中在 MAPK(mitogen-activated protein kinase)信號通路、細胞骨架調控、Rap1信號通路等(圖3)。
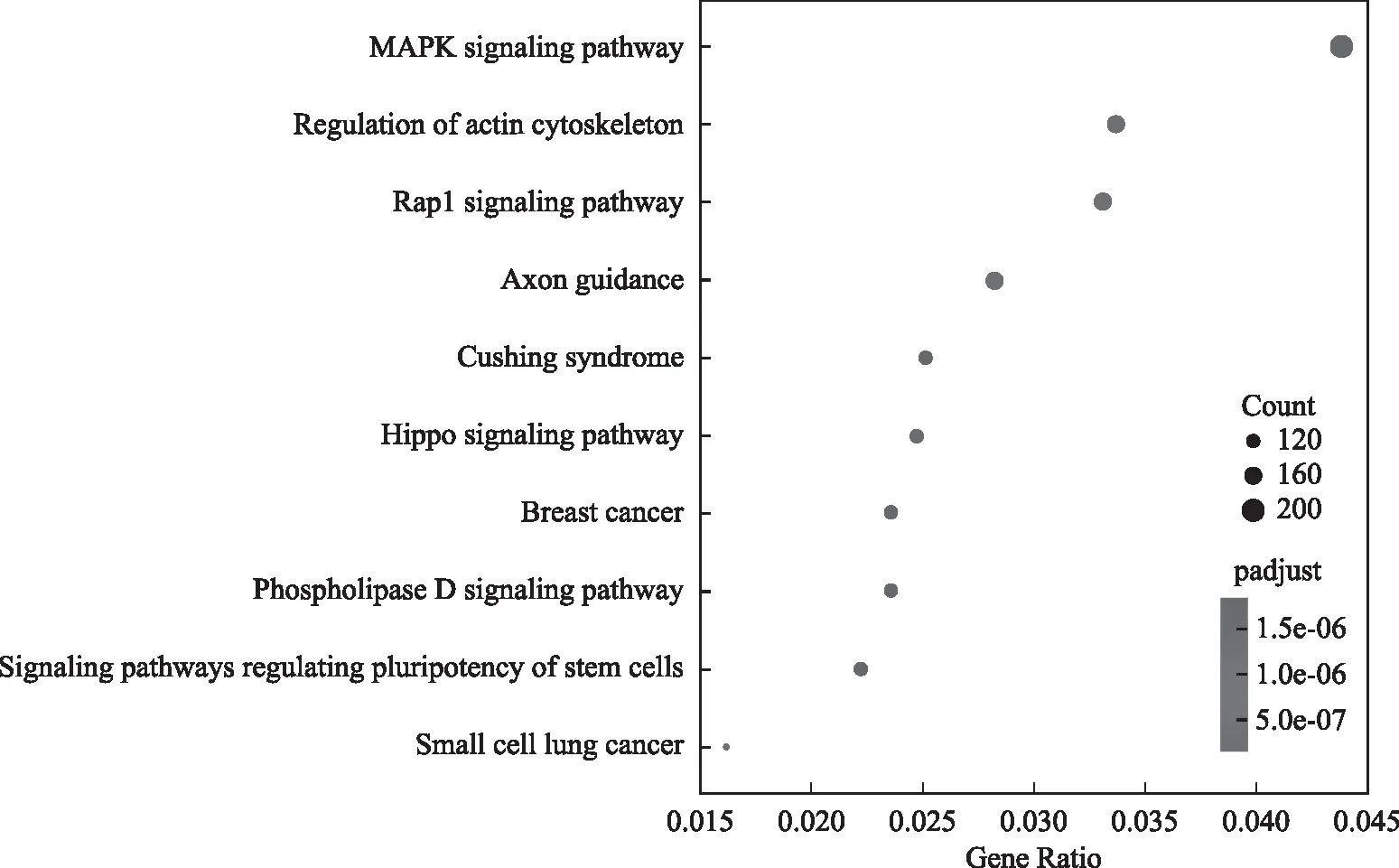
圖3 差異peak相關基因KEGG富集分析
討 論
目前現有的用于染色質開放區域捕獲的技術手段包括利用脫氧核糖核酸酶DNase Ⅰ酶切基因組尋找高敏位點的DNase-seq(DNaseⅠhypersensitive sites,DHSs);利用有機溶劑甲醛對染色體裸露的DNA進行固定的FAIRE-seq(Formaldehyde-Assisted Isolation of Regulatory Elements with Sequencing)。這些方法對于細胞數量要求甚高,而且實驗步驟繁瑣。本研究利用二氫葉酸還原酶抑制劑MTX誘導葉酸代謝障礙作用胚胎干細胞,采用ATAC-seq發現全基因組染色質開放程度即發生明顯變化,涉及基因尤以組織器官形成為主,提示葉酸代謝能夠影響早期胚胎發育表觀修飾調控。
2017《Development》通過ATAC-seq技術獲得早期小鼠胚胎組織的全基因組順式調控元件序列譜,并對預測增強子位點進行了功能鑒定,發現3個新的增強子序列對于小鼠神經板前后體軸的發育是必須的。通過分析motif,發現屬于Smad蛋白家族成員的motif序列[11]。轉錄因子信號轉導和轉錄激活因子-3(signal transducer and activator of transcription factor-3,STAT3)參與很多基因的表達與調控,并與其他轉錄因子形成復雜的網絡調控。Fujitani等[12]發現在哺乳動物前腦發育時期即有JAT/STAT蛋白表達,進一步檢測發現在妊娠14.0~18.0 d大鼠胚胎的大腦皮質、紋狀體、基底前腦和海馬都有STAT3的表達,而STAT3敲除的鼠胚胎發育至妊娠6.5 d就退化,提示STAT3對于胚胎早期的發育是必要的。對小鼠胚胎的研究發現,妊娠9.5 d時,STAT3在腦區有高水平的表達,此時間點恰好為神經系統發育的重要時期。推辭STAT3可能參與神經管閉合的關鍵信號通路調節[13]。ATAC-seq研究揭示STAT3通過結合染色質開放區域調控位點能夠與KLF6基因啟動子區形成loop環狀結構,協同促進神經突觸的發育與生長,決定中樞神經系統神經前體細胞的分化命運[14]。Fullard等[15]通過ATAC-seq技術建立了成人大腦(神經元,非神經元)的可接近性染色質圖譜,繪制了5個個體14個不同大腦區域的染色質開放性結構域,神經元較非神經元具有更高的染色質開放區,意味著此處有更多的順式調控元件存在,參與基因表達調控。紋狀體神經元會存在特殊的染色質開放區域,具有識別分子途徑和生物功能差異。通過motif分析,鑒定出大腦區域特異性蛋白編碼序列及長鏈非編碼RNA。本研究ATAC-seq證實葉酸代謝障礙下的mESC染色質可接近性與正常葉酸培養相比,其開放程度呈下降趨勢,提示調控因子結合位點發生改變,可能影響早期胚胎發育進程重編程。因此我們推測葉酸缺乏可能通過改變染色質可接近性影響組織器官命運分化,決定細胞命運分化,但其具體調控機制尚待進一步研究探討。
