油酸改性Fe2(MoO4)3用于稠油水熱催化降黏的研究
2019-07-08 06:15:48陸江銀云慶慶
石油煉制與化工 2019年7期
關鍵詞:催化劑
成 浪,李 玲,陸江銀,云慶慶
(1.新疆大學石油天然氣精細化工教育部重點實驗室,烏魯木齊 830046;2.新疆油田分公司實驗檢測研究院)
由于常規輕油資源的枯竭,在過去幾十年稠油已成為主要研究熱點[1]。然而,稠油黏度高、成分復雜,以經濟有效的方式開采、輸送稠油依然具有難度[2-3]。目前,循環蒸汽吞吐技術是最普遍且最有效的稠油開采技術,該技術是通過水蒸氣給油藏提供熱量從而達到稠油降黏目的[4-5]。Shokrlu等發現過熱蒸汽不僅可以降低稠油的黏度,而且還能與稠油發生反應,改變稠油的性質與組成,并將此化學反應描述為水熱裂解[6]。且在催化劑加入的情況下,水熱裂解反應的效率可以得到很大程度上的提高,稠油降黏效果更加顯著[7]。稠油水熱催化改質降黏可實現稠油不可逆降黏,該技術具有潛在的應用前景,是未來經濟高效的稠油開采技術[8]。
本研究用油酸對Fe2(MoO4)3進行改性,并用于新疆克拉瑪依稠油的降黏,考察催化劑用量、反應溫度、反應時間對稠油催化改質降黏的影響,通過稠油族組成分析、FT-IR、元素分析、1H NMR等分析手段對稠油降黏原因進行探討。
1 實 驗
1.1 主要試劑與設備
油酸、鉬酸銨、三氯化鐵、氫氧化鈉、四氫萘、正庚烷、正己烷、甲苯、乙醇、石油醚,分析純;三氧化二鋁(層析用,100~200目);稠油(來自新疆克拉瑪依,黏度(50 ℃)為13 200 mPa·s)。
CJF-0.2 L高溫高壓反應釜,NDJ-8S旋轉黏度計,Vario MACRO cube 元素分析儀,EQUINOX 50紅外光譜儀,BENFEN 3420氣相色譜儀,Bruker D8 advance X射線粉末衍射儀,Varian INOVA-400超導核磁共振波譜儀,SDT Q600 熱重分析儀。
1.2 催化劑OA-Fe2(MoO4)3的制備
將一定量的油酸(OA)與NaOH加入到500 mL單口燒瓶中,然后加入混合溶劑(蒸餾水、乙醇和正己烷體積比為1∶1∶4),升溫至60 ℃恒溫0.5 h。加入一定量的FeCl3·6H2O和(NH4)6Mo7O24·4H2O溶液,恒溫4 h后,用分液漏斗濾掉水層,有機層用蒸餾水清洗后轉入200 mL水熱釜中,在160 ℃下保持8 h,然后產物用乙醇及蒸餾水清洗,最后在65 ℃下干燥12 h得到催化劑,記為OA-Fe2(MoO4)3。
1.3 稠油催化改質降黏試驗
取50 g稠油置于高溫高壓反應釜中,加入一定量的水、供氫劑、催化劑后密封。通入N2,排出釜內空氣后將反應壓力升至3 MPa,將溫度升至反應溫度。反應一定時間后,停止反應。待反應釜冷卻至室溫后,取出油樣,在50 ℃下(下同)測定降黏稠油的黏度,分析稠油族組成變化情況。稠油降黏率計算式如下:
式中:η為稠油降黏率;μ0為稠油初始黏度;μ為稠油水熱催化降黏后的黏度。
1.4 表征與測試
族組成分析采用NB/SH/T 0509—2010方法;采用氣相色譜儀分析裂解氣組成;采用FT-IR分析自制催化劑、反應前后稠油及重組分的結構;采用元素分析儀分析稠油及重組分的C,H,N,S元素含量;采用XRD表征催化劑晶體結構;采用熱重方法分析催化劑的熱穩定性;采用1H NRM表征稠油及重組分的分子結構。
2 結果與討論
2.1 催化劑表征
2.1.1 FT-IR圖1為油酸和OA-Fe2(MoO4)3的FT-IR圖譜。從圖1可以看出:波數2 920 cm-1和2 851 cm-1處為甲基和亞甲基的吸收峰;波數744 cm-1處為MoO4八面體υ1、υ2的偶合,波數864 cm-1處為Mo—O鍵振動吸收峰[9],波數540 cm-1處為Fe—O鍵吸收峰;波數1 690 cm-1處的吸收峰表明催化劑含有C=O鍵;波數1 439 cm-1處是—COO—對稱拉伸吸收峰,表明催化劑中包含了—COO—基團[10-11]。因此,可以認為Fe2(MoO4)3與油酸存在強烈作用并形成復合物,是所預想的催化劑。
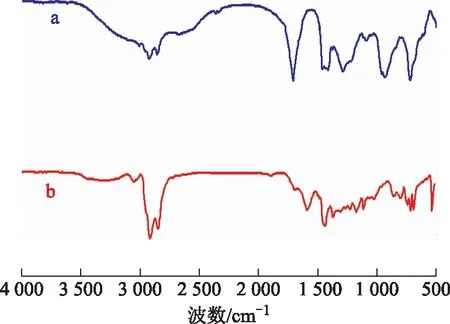
圖1 油酸和OA-Fe2(MoO4)3的FT-IR圖譜a—油酸; b—OA-Fe2(MoO4)3
2.1.2 XRD圖2為OA-Fe2(MoO4)3樣品的XRD圖譜。從圖2可以看出,OA-Fe2(MoO4)3樣品的XRD圖譜的特征衍射峰與3種Fe2(MoO4)3標準圖譜吻合度較高,說明所制備的催化劑是Fe2(MoO4)3。圖2中OA-Fe2(MoO4)3各特征衍射峰較為尖銳,雜峰較少,說明該催化劑純度高且相對結晶度高。
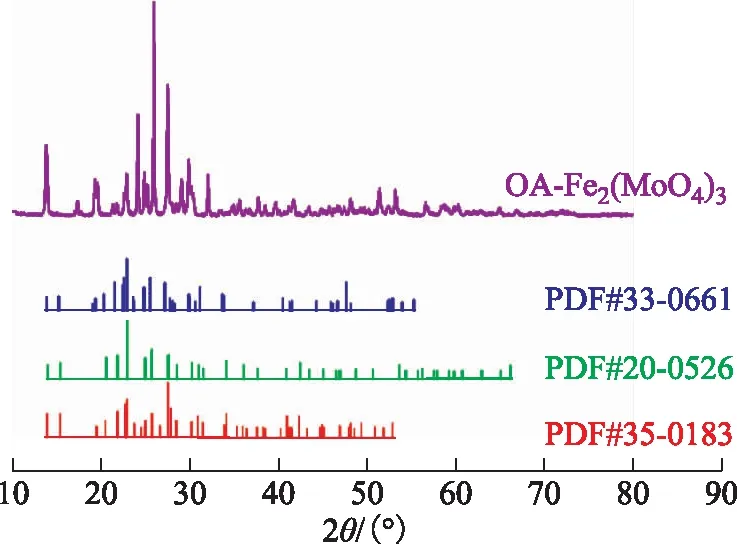
圖2 OA-Fe2(MoO4)3的XRD圖譜
2.1.3 熱重分析為了更好地了解所制備的催化劑的熱穩定性,對催化劑進行熱重分析,結果見圖3。樣品的熱重分析是從25 ℃到700 ℃,升溫速率為10 ℃/min,N2氣氛。從圖3可以看出,OA-Fe2(MoO4)3的失重曲線大致可以分成3個階段:第1階段25~250 ℃的失重可以歸因于結合水與有機溶劑的蒸發;第2階段250~430 ℃的失重可能是由于油酸不飽和碳鏈斷裂所致;第3階段430~593 ℃的失重可以認為是剩余油酸的蒸發。在593 ℃時,Fe2(MoO4)3的殘存質量約為83.31%。熱重分析結果表明制備的催化劑有良好的熱穩定性,可以用于稠油水熱催化改質降黏。
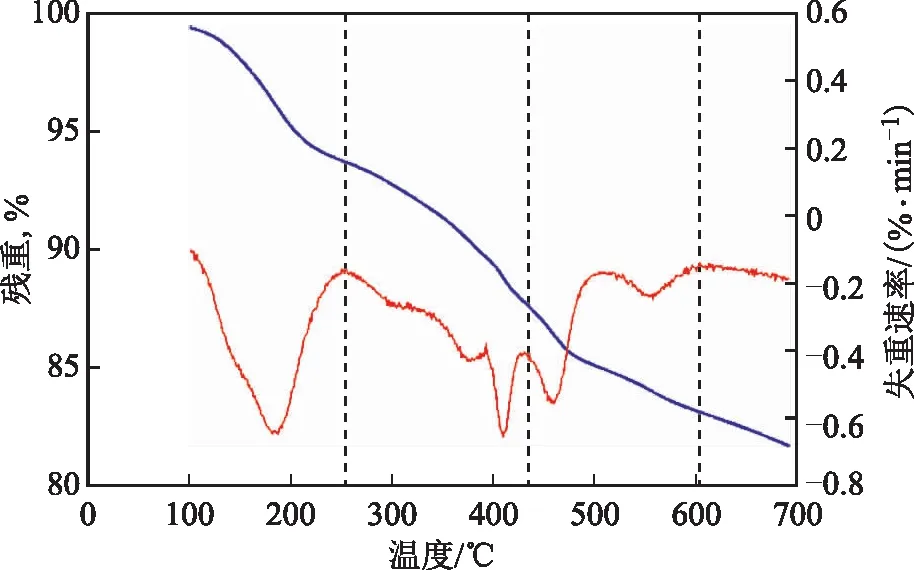
圖3 OA-Fe2(MoO4)3的熱重曲線 —殘重; —失重速率
2.2 催化劑和反應條件對稠油降黏的影響
2.2.1 催化劑的影響將50 g稠油和1 mL四氫萘加入到反應釜中,反應溫度控制在200 ℃,反應時間為12 h,考察催化劑的加入對稠油水熱催化改質降黏效果的影響,結果見圖4。其中,空白試驗油樣指不添加催化劑、水和四氫萘;水熱裂解油樣指僅添加水和四氫萘,其中油水質量比為7∶3;催化裂解油樣指僅添加催化劑及四氫萘,其中催化劑添加量(w,下同)為0.2%;水熱催化裂解油樣指添加了水、催化劑和四氫萘,其中油水質量比為7∶3,催化劑添加量為0.2%。
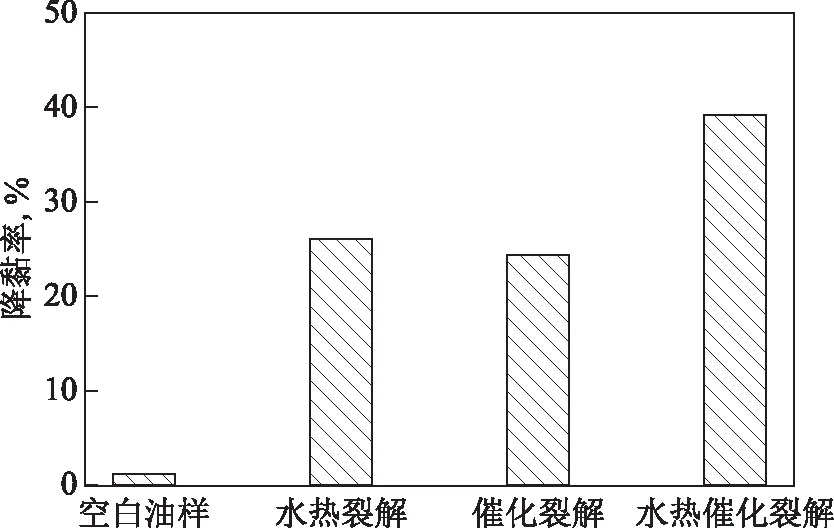
圖4 催化劑對稠油水熱裂解降黏效果影響
從圖4可以看出:水熱裂解、催化裂解對稠油均有降黏效果,降黏率分別為26.06%和24.30%;水熱催化裂解后的降黏效果最好,降黏率達到39.24%,表明催化劑的加入有助于改善稠油水熱裂解降黏效果,說明水與催化劑之間存在協同作用,兩者共同作用更有助于稠油降黏。
2.2.2 催化劑添加量的影響將50 g稠油、水(油水質量比為7∶3)、1 mL四氫萘和催化劑加入到反應釜中,反應溫度控制在200 ℃,反應時間為12 h,考察催化劑添加量對稠油水熱催化降黏效果的影響,結果見圖5。從圖5可以看出:隨催化劑添加量的增加,降黏效果隨之提高;催化劑添加量小于0.4%時,降黏效果變化趨勢明顯,當催化劑添加量為0.4%時,稠油黏度降至7 440 mPa·s,降黏率為43.63%;當添加量繼續增加至1.0%時,降黏效果變化趨勢較為平緩,此時稠油黏度降至7 060 mPa·s,降黏率為46.51%,與催化劑添加量為0.4%時相比僅變化了2.88百分點。因此,催化劑最佳添加量選擇0.4%。
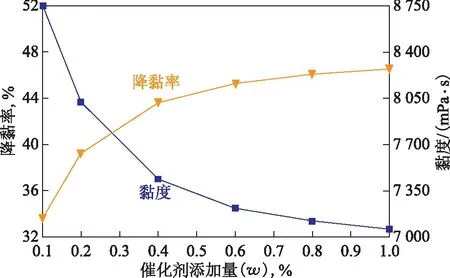
圖5 催化劑添加量對稠油水熱催化降黏效果的影響
2.2.3 反應溫度的影響水熱催化改質降黏過程中,需要滿足一定的能量要求才能使稠油分子中部分化學鍵發生斷裂[12]。因此,反應溫度是反應過程中必須考察的重要指標。將50 g稠油、水(油水質量比為7∶3)、1 mL四氫萘和質量分數0.4%的催化劑加入到反應釜中,反應時間為12 h,考察反應溫度對稠油水熱催化降黏效果的影響,結果見圖6。
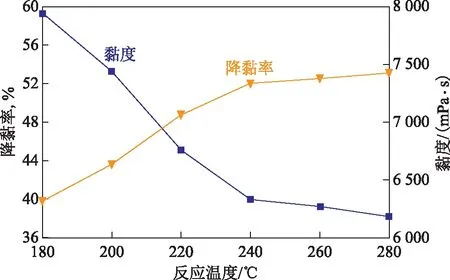
圖6 反應溫度對稠油水熱催化降黏效果的影響
從圖6可以看出:反應溫度對降黏率有著重要的影響,隨反應溫度的升高,稠油黏度逐漸降低,降黏率逐漸增加;當反應溫度為180~240 ℃時,稠油黏度從7 940 mPa·s降至6 430 mPa·s,降黏率從39.84%升至52.05%,說明該溫度區間是催化劑發揮催化性能的最佳溫度段;當溫度區間為240~280 ℃時,黏度變化緩慢,降黏率趨于平穩。考慮到油藏以及水蒸氣溫度,最佳反應溫度選擇為240 ℃。
2.2.4 反應時間的影響將50 g稠油、水(油水質量比為7∶3)、1 mL四氫萘和質量分數0.4%的催化劑加入到反應釜中,反應溫度控制在240 ℃,考察反應時間對水熱催化改質后稠油黏度的影響,結果見圖7。從圖7可以看出:反應時間在12~36 h區間,稠油黏度隨反應時間的增加而下降;在36 h之后,黏度下降趨勢減緩并趨于穩定;當反應時間為36 h時,稠油黏度降至4 900 mPa·s,此時降黏率為61.21%。反應時間超過36 h后,黏度降低幅度較小的原因可能是水熱催化改質過程中,膠質、瀝青質是主要的反應產物,其含量隨反應的進行逐步減少,反應速率也隨之降低[13],此時可認為改質降黏反應基本完成,故最佳反應時間選擇為36 h。

圖7 反應時間對稠油水熱催化降黏效果的影響
3 稠油水熱催化改質降黏原因分析
3.1 油樣族組成
表1為水熱催化改質降黏在最佳反應條件下(催化劑添加量為0.4%,反應溫度為240 ℃,反應時間為36 h)油樣與空白試驗油樣族組成變化情況。從表1可以看出:水熱催化改質后族組成發生了顯著變化;膠質和瀝青質質量分數分別從31.0%和13.2%降至20.7%和10.3%,分別降低10.3百分點和2.9百分點;飽和分和芳香分質量分數分別從32.9%和22.9%增至41.5%和27.5%,分別增加8.6百分點和4.6百分點。這表明水熱催化反應可以有效促進稠油中的重組分(膠質和瀝青質)裂解成輕組分(飽和分和芳香分)。
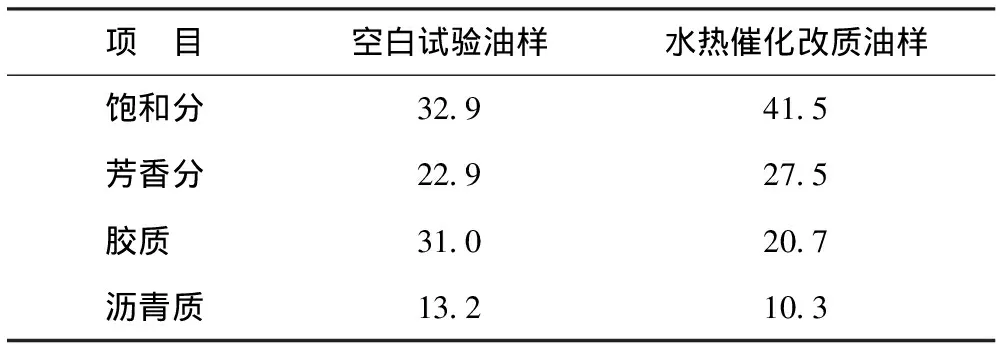
表1 空白試驗油樣與水熱催化改質油樣的族組成 w,%
3.2 裂解氣分析
對空白試驗和水熱催化改質后的氣相產物用氣相色譜儀進行成分及含量分析,結果見表2。從表2可以看出,反應裂解氣中主要含有CO、CO2、H2以及小分子烴類物質。其中,水熱催化改質后CO2含量增加明顯,說明稠油在水熱催化改質過程中發生了脫羧反應;而烷烴、烯烴等物質的含量增加,說明催化裂解過程中稠油中重組分發生了解聚和橋鍵斷裂,產生了小分子物質[14]。此外,C3和C4等組分含量增加則可能是自由基機理與碳正離子機理同樣適用于本反應體系,稠油在催化劑的作用下,大分子發生脫側鏈反應,進一步生成氣態烴等小分子[15]。
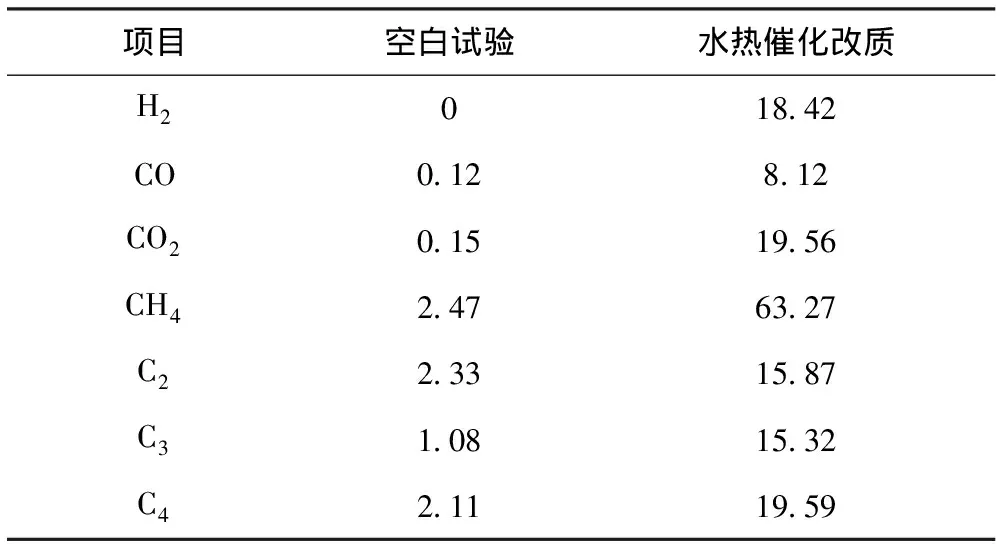
表2 空白試驗與水熱催化改質裂解氣組成 φ,%
注:表中數據以N2體積分數為100%基礎計算得到。
3.3 元素分析
表3為空白試驗油樣和水熱催化改質油樣的元素分析結果。從表3可以看出,油樣的N、S質量分數分別從改質前的0.59%和1.01%降至改質后的0.58%和0.83%,C、H質量分數分別從改質前的87.37%和11.03%變為改質后的86.88%和11.71%,H/C原子比從改質前的1.51增至改質后的1.62。通過比較可知,水熱改質后油樣的N、S質量分數分別降低0.01百分點和0.18 百分點,C質量分數降低0.49百分點,H質量分數增加0.68百分點,H/C原子比增加了0.11。水熱催化改質降黏后的油樣H/C原子比略增加的原因可能是稠油中不飽和組分加氫的結果。而且,不難發現,與空白試驗油樣相比,水熱催化改質后油樣中的N、S含量均有一定程度降低,但S含量降低程度更大,說明水熱催化裂解過程中C—S鍵、C—N鍵均發生了斷裂,同時也說明C—S鍵比C—N鍵更易斷裂。
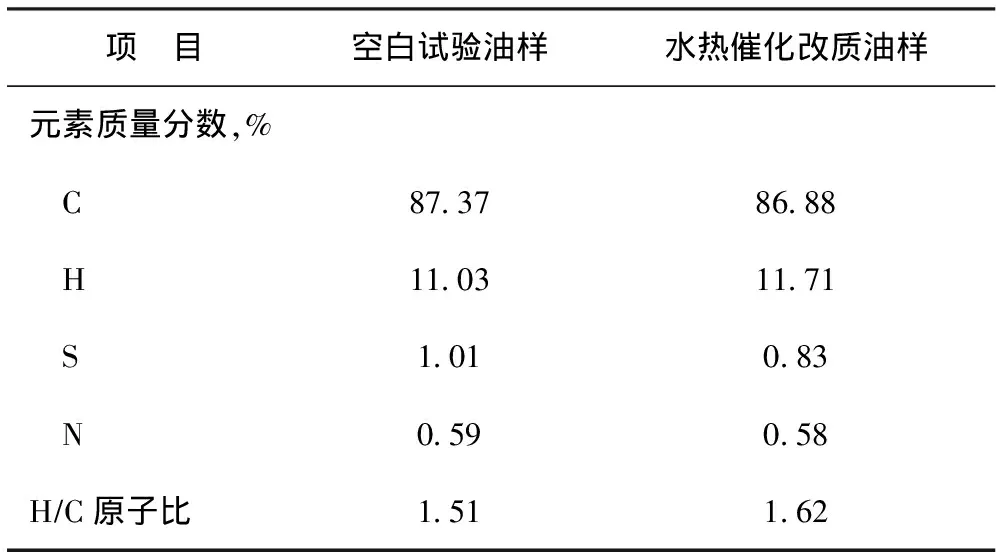
表3 空白試驗油樣與水熱催化改質油樣的元素含量
表4為空白試驗油樣與水熱催化改質油樣的重組分(膠質、瀝青質)元素分析結果。從表4可以看出,水熱催化改質后,膠質、瀝青質中的S、N含量均有所降低,H/C原子比均有所增加。其中S含量的降低進一步說明了在水熱催化改質過程中,C—S鍵確實發生了斷裂,解釋了稠油黏度降低的原因。兩個重組分H/C原子比均增加,表明反應過程中加氫反應是同時進行的。
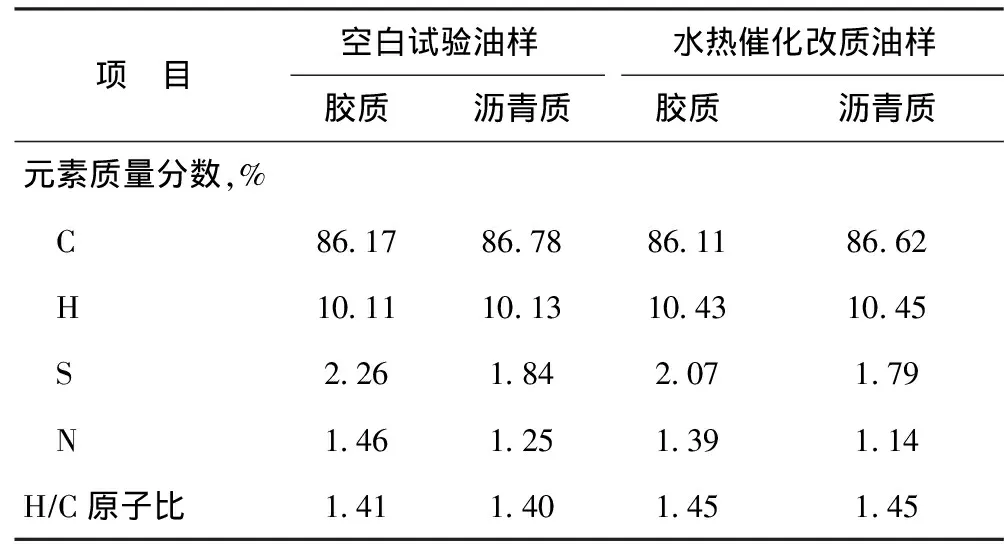
表4 空白試驗油樣與水熱催化改質油樣的膠質、瀝青質的元素含量
3.4 稠油、膠質和瀝青質的FT-IR分析
3.4.1 稠油的FT-IR分析圖8為空白試驗油樣與水熱催化改質油樣的FT-IR圖譜。從圖8可以看出:波數2 922 cm-1和2 853 cm-1處分別是甲基、亞甲基伸縮振動特征吸收峰;波數1 603 cm-1處的吸收峰為稠環芳烴特征峰,水熱催化改質后此峰增強,說明油樣中多環芳烴數減少較為明顯;波數1 454 cm-1和1 377 cm-1處分別是甲基、亞甲基彎曲振動特征吸收峰;波數742 cm-1處為長鏈烷烴(CH2)n(n≥4)特征吸收峰,水熱催化改質后此峰增強,說明反應產生了更多的長鏈烷烴,且烴類的飽和度增大。
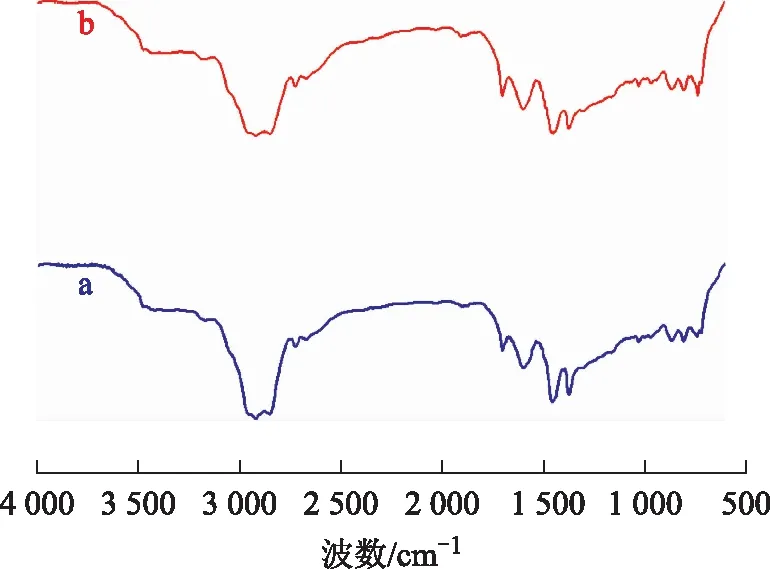
圖8 空白試驗油樣和水熱催化改質油樣的FT-IR圖譜a—空白試驗油樣; b—水熱催化改質油樣
3.4.2 膠質的FT-IR分析圖9為空白試驗油樣與水熱催化改質油樣的膠質的FT-IR圖譜。從圖9可以看出:波數2 920 cm-1和2 851 cm-1處是甲基、亞甲基伸縮振動吸收峰;波數1 454 cm-1和1 373 cm-1處是甲基、亞甲基彎曲振動吸收峰;波數1 678 cm-1處是膠質中的C=O吸收峰,水熱催化改質后此峰變弱,表明反應過程中發生了如C=O裂解的脫羧反應;波數1 597,1 373,864,812 cm-1處是膠質中的稠環芳烴吸收峰,水熱催化改質后峰面積變小,說明在反應過程中發生了開環反應,芳香環數減少;波數1 234,1 180,1 026 cm-1處的峰在改質反應后減弱,表明有C—S,C—O,C—N鍵斷裂。
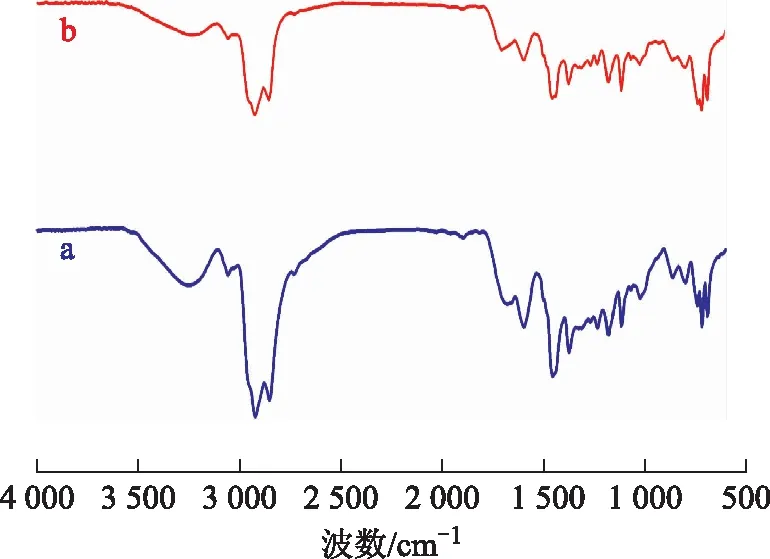
圖9 空白試驗油樣和水熱催化改質油樣膠質的FT-IR圖譜a—空白試驗油樣膠質; b—水熱催化改質油樣膠質
3.4.3 瀝青質的FT-IR分析圖10為空白試驗油樣與水熱催化改質油樣的瀝青質的FT-IR圖譜。從圖10可以看出:波數2 920 cm-1和2 851 cm-1處是甲基、亞甲基的特征吸收峰;波數1 504 cm-1處的峰在水熱催化改質后消失,說明存在部分脫烷基化反應[16];波數1 030,1 180,1 231 cm-1處的峰在水熱催化改質后變弱,表明有C—N,C—O,C—S鍵斷裂;波數802 cm-1處的峰在水熱催化改質后變弱,說明有脫烷基鏈反應發生;波數1 593 cm-1處的峰在水熱催化改質后增強,表明水熱催化裂解過程中油樣的芳香環數降低。
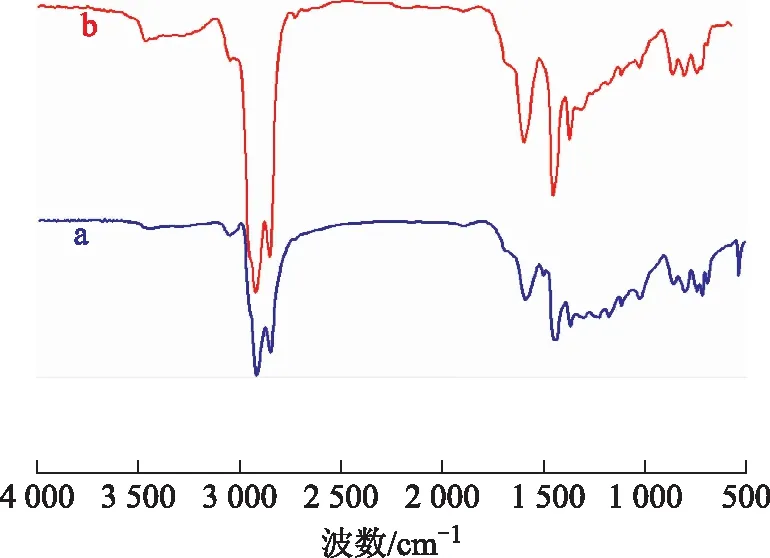
圖10 空白試驗油樣和水熱催化改質后油樣瀝青質的FT-IR圖譜a—空白試驗油樣瀝青質; b—水熱催化改質油樣瀝青質
膠質、瀝青質的FT-IR譜圖說明其主要是由稠環芳烴、烷基鏈以及雜原子構成,所有變化情況都說明在水熱催化改質降黏后,C—N,C—O,C—S鍵均有不同程度的斷裂,這一結果與元素分析結果一致。
3.5 膠質與瀝青質的1H NMR分析
油樣的膠質與瀝青質的1H NMR分析中以氘代氯仿為溶劑、四甲基硅烷為內標物。圖11為空白試驗油樣和水熱催化改質后油樣的膠質和瀝青質的1H NMR圖譜。
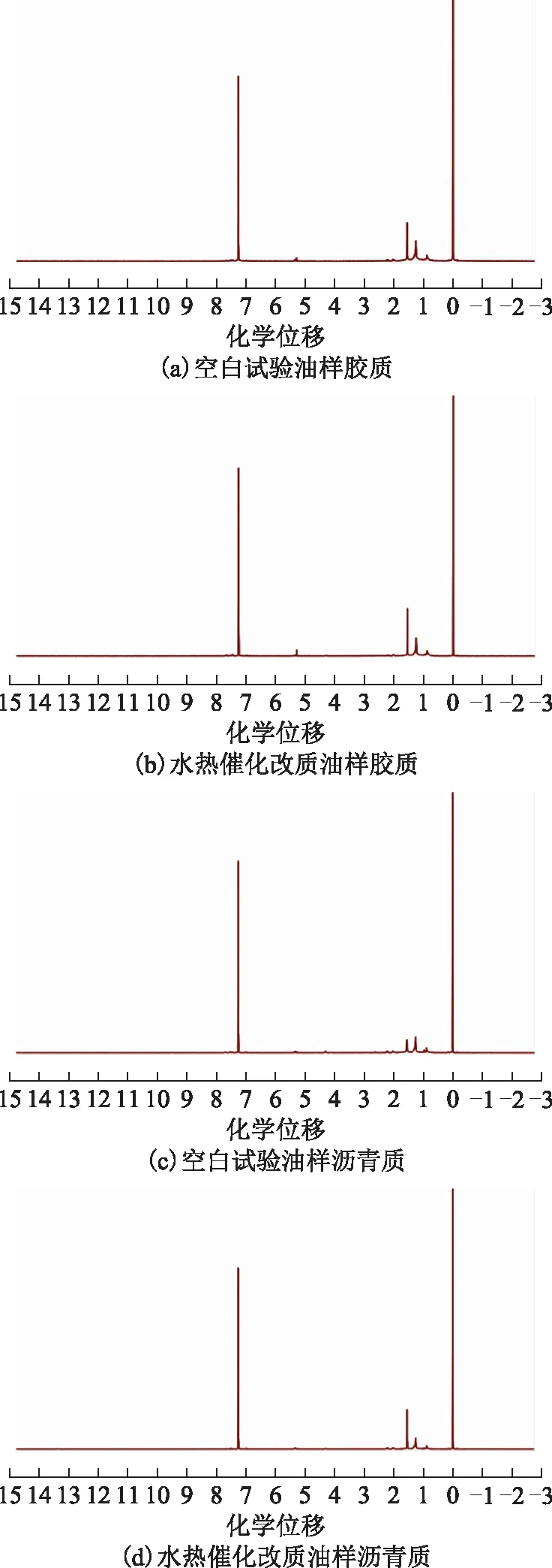
圖11 空白試驗油樣和水熱催化改質后油樣的膠質和瀝青質的1H NMR圖譜
表5為根據圖11中質子氫歸屬得到的結構參數[17],其中:HA為與芳香碳直接相連的氫原子數比例;Hα為與芳香環的α碳相連的氫原子數比例;Hβ為與芳香環的β碳上以及β以遠的—CH2、—CH基上的氫原子數比例;Hγ為與芳香環的γ碳相連的—CH3基上的氫原子數比例。從表5可以看出,反應后膠質和瀝青質的芳香度(fA)和芳香環系的縮合度參數(HAU/CA)降低。不飽和組分的加氫反應使得芳香性降低;芳香環系的縮合度參數的增加,可能是在水熱催化裂解過程中發生了開環加氫反應,多環芳烴分子轉化為重組分的小碎片所致[3]。并且,在反應后膠質、瀝青質支化度(BI)指數均降低,表明在改質后膠質、瀝青質結構中的支鏈數減少,這可能是大分子中部分側鏈發生斷裂,使得膠質、瀝青質相對分子質量降低,從而降低了稠油黏度[18]。
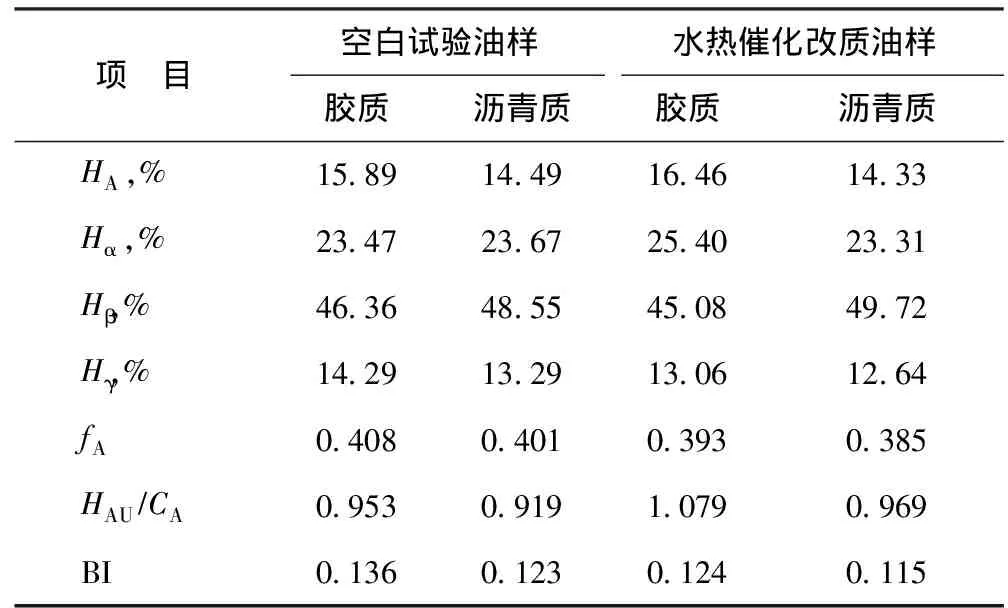
表5 空白試驗油樣與水熱催化改質油樣的膠質與瀝青質的1H NMR結構參數
4 結 論
通過水熱法成功合成出稠油水熱催化改質降黏催化劑OA-Fe2(MoO4)3,并用于新疆克拉瑪依稠油降黏試驗,得出水熱催化改質降黏試驗的最佳工藝條件為:OA-Fe2(MoO4)3添加量0.4%,反應溫度240 ℃,反應時間36 h,此時降黏率為61.21%。在反應過程中,重組分(膠質、瀝青質)通過C—N,C—O,C—S鍵的斷鍵,長鏈結構的斷裂以及不飽和組分的加氫轉變成輕組分(飽和分、芳香分),這可以從族組成、元素組成、fA及HAUCA得到驗證,這解釋了稠油黏度降低的原因,可為以后進一步的研究提供參考。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
