66例不明原因智力障礙/發(fā)育遲緩兒童臨床和遺傳學(xué)分析
2019-07-25 11:44:50洪曉文陳燕惠
福建醫(yī)科大學(xué)學(xué)報(bào) 2019年3期
關(guān)鍵詞:檢測(cè)
洪曉文, 陳燕惠
智力障礙(intellectual disability, ID)/發(fā)育遲緩(developmental delay, DD)是兒科發(fā)育行為門診常見的疾病。智力水平明顯低于同齡正常兒童,并有明顯的社會(huì)適應(yīng)困難者稱為智力發(fā)育障礙,對(duì)年齡>5歲的患兒診斷為ID,對(duì)年齡<5歲的幼兒更多使用DD[1-2]。據(jù)統(tǒng)計(jì),ID/DD的患病率為1%~3%[3],主要病因?yàn)檫z傳綜合征和染色體異常。但由于ID/DD的臨床癥狀重疊交叉且伴多種先天性畸形,無(wú)特異性,相關(guān)病因復(fù)雜且具有高度異質(zhì)性,臨床上不易診斷,染色體微陣列分析(chromosome microarray analysis,CMA)技術(shù)輔以全外顯子組基因測(cè)序(whole exome sequencing, WES)有助于對(duì)患兒的遺傳學(xué)病因進(jìn)行診斷。筆者收集2014年1月-2017年8月就診的不明原因的ID/DD患兒66例,分析總結(jié)患兒的臨床表現(xiàn)和基因測(cè)定結(jié)果,探討CMA結(jié)合WES在ID/DD診斷中的應(yīng)用,以期為ID/DD的臨床診斷提供更多的理論依據(jù)。
1 對(duì)象與方法
1.1對(duì)象 66例中,男性38例,女性28例,年齡1月~7歲。臨床癥狀或體征至少符合以下一點(diǎn):(1)存在智力低下、生長(zhǎng)發(fā)育遲緩;(2)具有異常面容;(3)伴有體表或內(nèi)臟畸形,如先天性心臟病等。同時(shí)排除以下情況:產(chǎn)傷、出生后中樞神經(jīng)系統(tǒng)感染或頭顱損傷、其他已知遺傳綜合征(如21三體綜合征等)以及常見遺傳代謝性疾病等,以及不同意進(jìn)行基因檢測(cè)和失訪的病例。
1.2方法
1.2.1收集資料 收集患兒的臨床病歷資料、智力測(cè)評(píng)報(bào)告、神經(jīng)影像學(xué)資料及視頻腦電圖檢查結(jié)果等。
1.2.2染色體微陣列分析 采用CytoScan基因芯片(CytoScanHD/CytoScan750K,美國(guó)Affymetrix公司)進(jìn)行全基因組范圍掃描,利用染色體分析軟件(Version:2.0.0.195,美國(guó)Affymetrix公司)進(jìn)行分析,檢測(cè)拷貝數(shù)變異(copy number variation, CNV)、長(zhǎng)期連續(xù)綿延純合子及嵌合體(≥20%)。
1.2.3遺傳學(xué)全外顯子測(cè)序 應(yīng)用過柱法從檢樣中提取DNA,通過高通量測(cè)序技術(shù),對(duì)基因外顯子區(qū)進(jìn)行直接測(cè)序,與參考序列(hg19)進(jìn)行比較,從而發(fā)現(xiàn)可能存在的基因突變。
2 結(jié) 果
2.1 臨床分析 (1)66例患兒均表現(xiàn)為智力低下,存在2個(gè)以上對(duì)應(yīng)年齡階段發(fā)育里程碑事件顯著延遲,其中具有孤獨(dú)癥譜系障礙樣患兒占50%。(2)檢出致病性變異患兒22例,男性10例,女性12例;1~11月6例,1~3歲12例,4~7歲4例。(3)致病性變異患兒22例中,伴有反復(fù)癲癇發(fā)作5例,有特殊面容、貫通掌、動(dòng)脈導(dǎo)管未閉、室間隔缺損、隱睪等先天性畸形15例。行頭顱MR檢查19例,其中9例有腦室擴(kuò)大、腦外間隙增寬等異常影像學(xué)特征;行視頻腦電圖檢查18例,其中12例發(fā)現(xiàn)背景慢波活動(dòng)增多,尖波、尖慢復(fù)合波、棘波、棘慢復(fù)合波等異常腦電圖表現(xiàn);19例行發(fā)育評(píng)估,提示輕到重度發(fā)育落后,結(jié)果為輕、中、重度智力低下分別為2,8,9例(表1)。
2.2遺傳學(xué)分析 66例患兒中,遺傳變異45例(68.2%),其中22例為致病性突變(包括20個(gè)臨床致病性CNV和4個(gè)致病基因突變),病因診斷明確,診斷陽(yáng)性率為33.3%(表2)。共對(duì)36例患兒進(jìn)行CNV分析,18例檢出臨床致病CNV,陽(yáng)性率為50%。Prader-Willi綜合征所占比例最高(4/18),其次為Williams-Beuren綜合征(3/18)。共對(duì)30例患兒進(jìn)行遺傳學(xué)WES,4例檢出臨床致病的基因突變,陽(yáng)性率為13.3%(表3)。
3 討 論
根據(jù)精神疾病診斷和統(tǒng)計(jì)手冊(cè),ID/DD患兒存在明顯的認(rèn)知功能障礙和適應(yīng)功能缺陷[4],患各種并發(fā)癥的風(fēng)險(xiǎn)增加。據(jù)文獻(xiàn)報(bào)道,17.4%~47.1%的ID/DD可以通過遺傳原因來解釋[5-6],本組33.3%的診斷率與之相吻合。目前,在遺傳因素中染色體異常占25%[7],21三體綜合征是最常見的染色體異常。已知單基因病例達(dá)10%,脆性X綜合征是與ID/DD相關(guān)的最常見的單基因缺陷病,約占智力殘疾病例的5%[8]。不明原因的ID/DD患者,尤其是臨床表型不具特異性及無(wú)明顯先天畸形和特殊面容的患兒,診斷十分困難,易漏診甚至誤診。近來,國(guó)內(nèi)外已有大量研究將CMA和WES應(yīng)用于不明原因ID/DD的遺傳學(xué)病因診斷,美國(guó)和歐洲指南指出,基因檢測(cè)是ID/DD必不可少的標(biāo)準(zhǔn)化診斷流程之一[9]。盡管CMA是目前不明原因ID/DD的一線診斷方法,但該技術(shù)也存在很大局限性,比如該技術(shù)不能檢測(cè)平衡易位、倒置,也無(wú)法對(duì)點(diǎn)突變致病的患者作出準(zhǔn)確的檢測(cè)。而WES彌補(bǔ)了該技術(shù)的不足,可以精確到單個(gè)基因位點(diǎn)的改變,為單基因ID/DD的診斷提供強(qiáng)有力的工具。
本研究階段,在筆者醫(yī)院神經(jīng)發(fā)育門診就診的ID/DD患兒共有350例,其中有明確原因的ID/DD患者共有184例,具體原因如下:感染性智力障礙(顱內(nèi)感染后遺癥如流腦、乙型腦炎)、中毒性智力障礙(如高膽紅素血癥腦病)、內(nèi)分泌功能障礙相關(guān)性智力障礙(如甲狀腺功能減退癥)、顱腦畸形相關(guān)性智力障礙(如腦回畸形等)、精神障礙相關(guān)性智力障礙(如孤獨(dú)癥譜系障礙等)、腦外傷及其他損傷相關(guān)性智力障礙(如顱內(nèi)出血、窒息等),主要通過出生史、新生兒疾病篩查、血氨基酸、尿有機(jī)酸、影像學(xué)、腦電圖和生長(zhǎng)發(fā)育史進(jìn)行明確,再排除不同意進(jìn)行基因檢測(cè)和失訪的病例。本研究最后根據(jù)患兒是否伴有先天畸形、特殊面容選擇CMA對(duì)36例不明原因ID/DD患兒進(jìn)行檢測(cè),為約50%的患兒做出了明確的分子遺傳學(xué)病因診斷。例1~5檢出15q11-q13缺失,此區(qū)域缺失與Prader-Willi綜合征或Angelman綜合征[10]。例1、例2、例4、例5均表現(xiàn)為智力發(fā)育遲緩,廣泛性發(fā)育落后于正常同齡兒。其中例1、例2、例5伴有癲癇發(fā)作;例2伴有先天性動(dòng)脈導(dǎo)管未閉。例3患兒出生后左側(cè)肢體張力低,少哭,哭聲低,體檢可見耳位低,雙手貫通掌。這5例患兒臨床表現(xiàn)與既往文獻(xiàn)報(bào)道有相似之處。前者可表現(xiàn)為嬰兒期發(fā)育遲緩,肌張力減退,喂養(yǎng)困難和低體質(zhì)量[11-12]。后者主要影響神經(jīng)系統(tǒng),表現(xiàn)為發(fā)育遲緩,智力低下,共濟(jì)失調(diào),癲癇發(fā)作和睡眠障礙,常有快樂的表情[13]。例6~8檢出7q11.23區(qū)域缺失,Williams-Beuren綜合征臨床表現(xiàn)為輕度到中度智力低下,特殊“elfin面容”(前額寬,眶周豐滿,鼻梁低平,顴骨低,人中長(zhǎng),唇厚嘴寬,牙齒發(fā)育不良),心血管功能異常(主動(dòng)脈瓣或肺動(dòng)脈瓣狹窄),其他癥狀包括癲癇、消化系統(tǒng)、泌尿生殖系統(tǒng)異常等[14]。例6~8皆表現(xiàn)為運(yùn)動(dòng)、言語(yǔ)發(fā)育落后于同齡兒,體格檢查發(fā)現(xiàn)患兒鼻翼較扁,手足較短,符合典型特殊“elfin面容”。例8患兒還表現(xiàn)為X形腿,右手貫通掌。即使本組患兒病史中暫無(wú)相關(guān)的心血管功能異常的臨床表現(xiàn),但是上述文獻(xiàn)表明,確實(shí)有必要進(jìn)行心血管功能監(jiān)測(cè)。目前,用于單基因ID/DD的二代測(cè)序主要有WES,但因檢測(cè)成本高,目前還未得到廣泛應(yīng)用。本研究針對(duì)不伴有明顯特殊面容或先天畸形的30例ID/DD患兒進(jìn)行WES,發(fā)現(xiàn)4例患兒存在明確單基因突變。例19和例20患兒MEF2C基因新生突變,診斷為智力障礙20型,呈常染色體顯性遺傳,臨床表型為刻板運(yùn)動(dòng)、癲癇、精神發(fā)育遲滯或腦部畸形[15]。MEF2C位于5q14.3,該基因的突變和缺失與嚴(yán)重的認(rèn)知障礙、刻板的運(yùn)動(dòng)、癲癇和大腦畸形有關(guān)[16]。例19和例20患兒均以“反復(fù)癲癇發(fā)作”為主訴就診,臨床表現(xiàn)為嚴(yán)重智力低下、言語(yǔ)不清以及輕微異常的面部特征,與既往文獻(xiàn)報(bào)道一致。可見,由于臨床異質(zhì)性,即使同一基因片段缺失或重復(fù)、同一基因突變患兒的臨床表型也有一定差異,甚至部分ID/DD患兒常常伴有孤獨(dú)癥譜系障礙樣、注意缺陷多動(dòng)障礙樣以及其他神經(jīng)發(fā)育障礙等異常行為,這給臨床診斷帶來挑戰(zhàn)。
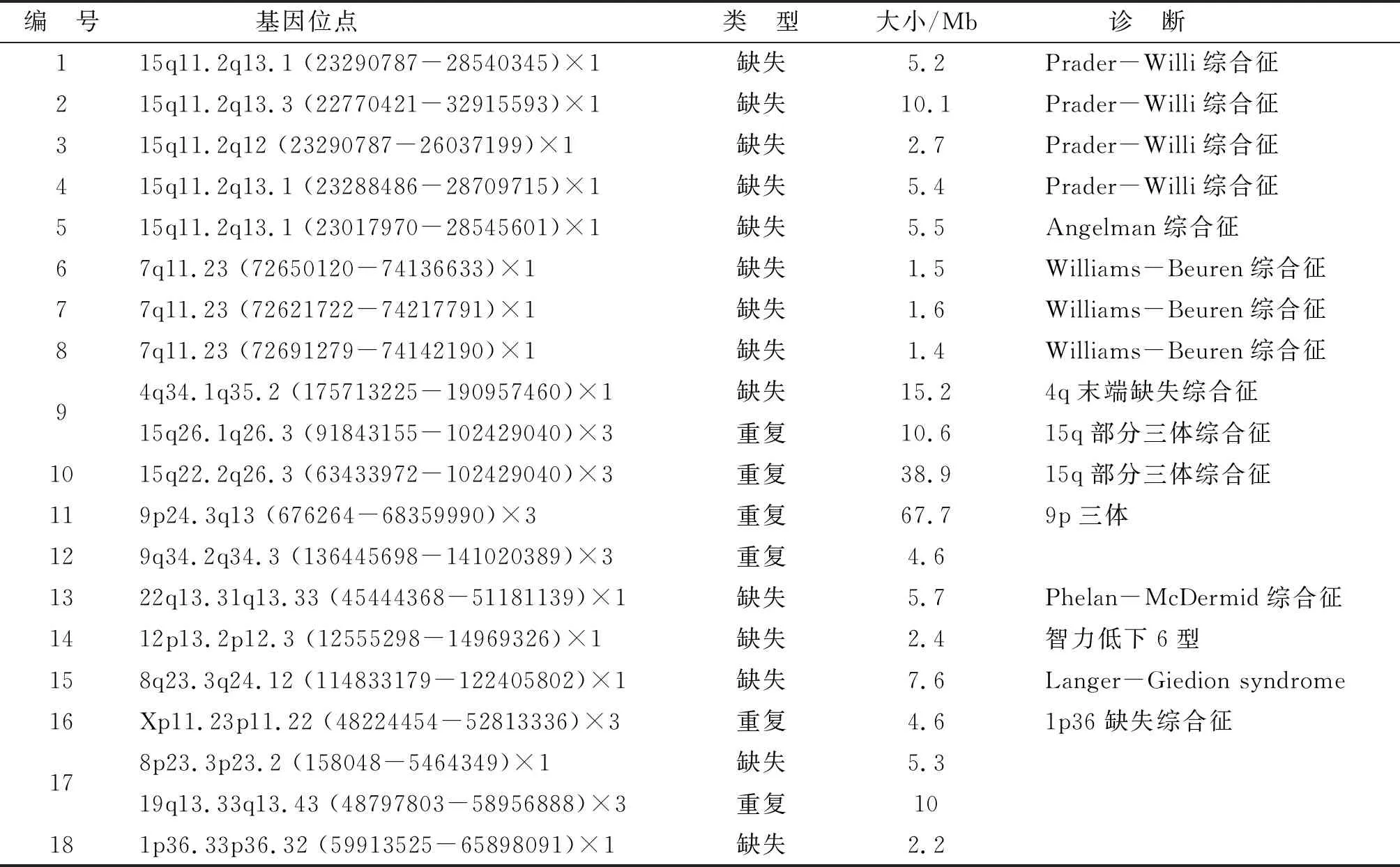
表2 18例檢出致病性CNV遺傳學(xué)分析

表3 4例檢出致病基因遺傳學(xué)分析
綜上所述,本研究應(yīng)用CMA和WES技術(shù)對(duì)原因不明的ID/DD患兒進(jìn)行檢測(cè),以期提高病因診斷率。但鑒于經(jīng)濟(jì)效益,并未按照最理想的診斷流程進(jìn)行檢測(cè)(首先進(jìn)行染色體核型分析,然后依次進(jìn)行CMA和WES,對(duì)檢測(cè)出的陽(yáng)性結(jié)果使用另一種實(shí)驗(yàn)方法驗(yàn)證)。研究表明,CMA與WES檢測(cè)陽(yáng)性病例與陰性病例在臨床表型、發(fā)病年齡等方面不存在明顯差別,另外CNV改變的ID/DD患兒與單基因突變的患兒在臨床表型等方面也不存在顯著差別[17]。盡管目前CMA是首選的檢查手段,相信隨著WES使用經(jīng)驗(yàn)的積累和ID/DD新的候選基因不斷被發(fā)現(xiàn),CMA結(jié)合WES將為臨床診斷不明原因的ID/DD提供更可靠的診斷結(jié)果。
猜你喜歡
中國(guó)設(shè)備工程(2022年12期)2022-07-11 04:33:00
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:36
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:34
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:50
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:48
