添加順序?qū)Ζ?乳球蛋白與EGCG及葡萄糖三元復(fù)合物結(jié)構(gòu)和功能的影響
2019-07-26 08:24:44姚文俊付珊琳鐘俊楨劉成梅
食品科學(xué) 2019年14期
姚文俊,周 磊,付珊琳,鐘俊楨*,劉成梅
(南昌大學(xué) 食品科學(xué)與技術(shù)國家重點(diǎn)實(shí)驗(yàn)室,江西 南昌 330047)
β-乳球蛋白(β-lactoglobulin,β-LG)是牛奶中乳清蛋白的主要成分,約占50%[1]。β-LG在中性pH值下為單體和二聚體共存狀態(tài)。每個單體由162 個氨基酸殘基組成,分子質(zhì)量約為18.4 kDa[2]。它含有8 個反平行β-折疊和α-螺旋[3]。β-LG是一種優(yōu)良的天然活性載體[4],它可以結(jié)合多種活性物質(zhì),包括親水性、疏水性和兩親性物質(zhì)。
食物體系是一個多重復(fù)雜的系統(tǒng)。目前,大量研究報道了β-LG和其他活性物質(zhì)的組合對其結(jié)構(gòu)和功能的影響。最常見的方法有2 種,一種是直接混合以氫鍵、疏水鍵為主要作用力形成非共價復(fù)合物[5]。Li Min等[6]通過熒光等光學(xué)手段研究了牛血清白蛋白與表沒食子兒茶素沒食子酸酯(epigallocatechin gallate,EGCG)間的非共價結(jié)合。姜黃素與β-LG通過疏水相互作用結(jié)合后,蛋白質(zhì)結(jié)構(gòu)發(fā)生變化,姜黃素的抗氧化活性得到改善[7]。然而,由疏水鍵、氫鍵等形成的非共價復(fù)合物易受溶液和環(huán)境的影響而發(fā)生可逆現(xiàn)象[8-9]。一種是以堿處理、酶催化、自由接枝聚合、美拉德反應(yīng)等方法共價結(jié)合形成共價復(fù)合物,例如Wu Xuli等[10]通過自由基法得到β-LG與EGCG和綠原酸(chlorogenic acid,CA)共價復(fù)合物,發(fā)現(xiàn)β-LGEGCG和β-LG-CA可以降低β-LG的致敏性。蛋白質(zhì)與其他物質(zhì)形成共價復(fù)合物是當(dāng)前的研究熱點(diǎn)。通過共價處理形成的共價鍵是不可逆的,且其復(fù)合物更穩(wěn)定[11-12]。特別地,共價形成的復(fù)合物較非共價復(fù)合物具有更好的結(jié)構(gòu)和功能特性。β-LG與兒茶素通過自由基接枝法得到的共價復(fù)合物與非共價復(fù)合物對比發(fā)現(xiàn),共價復(fù)合物的抗氧化性和對β-胡蘿卜素的保留率有所提高[13]。乳清分離蛋白與EGCG通過堿法得到共價復(fù)合物后,起泡性和乳化性都得到改善[14]。牛血清白蛋白和葡聚糖的共價復(fù)合物可以抑制牛血清白蛋白的聚集,而二級結(jié)構(gòu)不受美拉德反應(yīng)時間的影響[15]。但是,大量的研究都集中在2 種物質(zhì)的結(jié)合上,例如蛋白質(zhì)和多酚,蛋白質(zhì)和多糖以及其他二元復(fù)合物,目前關(guān)于β-LG與多酚和多糖的三元共價復(fù)合物的影響研究較少。
近年來,三元復(fù)合物成為研究的熱點(diǎn),三元復(fù)合物較二元復(fù)合物會展現(xiàn)更多不同的結(jié)構(gòu)功能性質(zhì)。如Harbertson等[16]發(fā)現(xiàn)單糖和雙糖可以形成氫鍵增加單寧與蛋白質(zhì)之間的作用力,并提高蛋白質(zhì)單寧復(fù)合物的溶解度。多糖還可以競爭抑制蛋白質(zhì)多酚復(fù)合物的聚集[17]。此外,多酚也會對蛋白質(zhì)和多糖的復(fù)合物產(chǎn)生影響。槲皮素可通過靜電和疏水相互作用增加阿拉伯膠與β-LG的相互作用[18]。因此,多糖對蛋白質(zhì)-多酚復(fù)合物和多酚對蛋白質(zhì)-多糖復(fù)合物有不同的影響。對于特定的蛋白質(zhì)多酚多糖,不同的添加順序?qū)⒂绊憦?fù)合物之間的結(jié)構(gòu)和功能性質(zhì)。Yang Wei等[19]以不同的添加順序獲得乳鐵蛋白,果膠和EGCG三元非共價復(fù)合物。發(fā)現(xiàn)EGCG和果膠的添加順序在最終形成的復(fù)合物的結(jié)構(gòu)和功能方面差異很大。然而,目前大多數(shù)研究都集中在非共價復(fù)合物上。對于不同添加順序?qū)θ矁r結(jié)合復(fù)合物結(jié)構(gòu)的影響以及與非共價復(fù)合物的區(qū)別尚不清晰。
因此,本研究采用2 種方法形成不同添加順序的β-LG、EGCG和葡萄糖(glucose,Glc)共價復(fù)合物和非共價復(fù)合物。通過熒光光譜和圓二色(circular dichroism,CD)光譜表征其結(jié)構(gòu)變化,并比較它們的起泡性和乳化性質(zhì)。探討不同添加順序?qū)?fù)合物的影響以及共價復(fù)合物與非共價復(fù)合物的區(qū)別,為食品加工順序?qū)κ称敷w系的影響研究提供理論參考。
1 材料與方法
1.1 材料與試劑
牛乳β-LG、EGCG 美國Sigma公司;8-苯胺基-1-萘磺酸鈉(8-anilino-1-naphthalenesulfonic acid,ANS)、十二烷基硫酸鈉(sodium dodecyl sulfate,SDS)、丙烯酰胺、過硫酸氨(ammonium persulphate,AP)、Glc 北京索萊寶生物科技有限公司;其他試劑均為國產(chǎn)分析純。
1.2 儀器與設(shè)備
FreeZone 12-Plus真空冷凍干燥機(jī) 美國Labconco公司;Ultra-TurraxT25高速分散機(jī) 德國IKA公司;UV-1600PC紫外分光光度計(jì) 上海美譜達(dá)儀器有限公司;F-7000熒光分光光度計(jì) 日本Hitachi公司;Mini-Protean Tetra Cell電泳儀 美國Bio-Rad公司;MOS-450 CD光譜儀 法國Bio-Logic公司。
1.3 方法
1.3.1 共價復(fù)合物及非共價復(fù)合物的制備
參考Rawel等[20]的方法,采用堿法合成β-LG-EGCG共價復(fù)合物。0.25 g β-LG溶解在25 mL去離子水中攪拌過夜,然后用0.1 mol/L NaOH溶液將蛋白質(zhì)溶液的pH值調(diào)節(jié)至9.0。稱取4 mg EGCG溶解在25 mL去離子水中,使其濃度為0.35 mmol/L,并將pH值調(diào)至9.0。蛋白質(zhì)溶液與EGCG溶液按體積比1∶1混合并連續(xù)攪拌24 h(加入質(zhì)量分?jǐn)?shù)0.02%的疊碳化鈉)。然后透析48 h除去多余的多酚。最后,冷凍干燥得到樣品。
參考Liu Fuguo等[21]的方法,采用美拉德反應(yīng)制備β-LG-EGCG-Glc共價復(fù)合物,稱取β-LG-EGCG共價復(fù)合物凍干樣品與Glc分別溶解在去離子水中,質(zhì)量濃度為20 mg/mL,等比例混合后用0.1 mol/L的HCl溶液調(diào)節(jié)pH值至7.0,冷凍干燥得到凍干樣品后放入裝有飽和溴化鉀溶液的干燥器中(60 ℃,相對濕度79%)反應(yīng)24 h得到β-LG-EGCG-Glc共價復(fù)合物。
按照上述方法,按不同的順序制備β-LG-Glc-EGCG 共價復(fù)合物。非共價復(fù)合物的制備除去共價復(fù)合物制備過程中的堿法和美拉德反應(yīng)外,其余步驟與上述方法相同。將β-LG-EGCG-Glc共價復(fù)合物命名為β-LGEGCG-Glc con,β-LG-Glc-EGCG共價復(fù)合物命名為β-LGGlc-EGCG con;β-LG-EGCG-Glc非共價復(fù)合物命名為β-LG-EGCG-Glc mix,β-LG-Glc-EGCG非共價復(fù)合物命名為β-LG-Glc-EGCG mix。
1.3.2 SDS-PAGE分析
SDS-聚丙烯酰胺凝膠電泳(polyacrylamide gelelectrophoresis,PAGE)根據(jù)Laemmli等[22]的方法并略作改動。使用濃縮膠5%、分離膠12%。首先恒定電壓80 V維持15 min后換成恒定電壓200 V維持45 min。每個孔的上樣量為10 μL。電泳之后,通過考馬斯亮藍(lán)R-250對凝膠條帶進(jìn)行染色后再用脫色液脫色。采用低分子質(zhì)量的蛋白質(zhì)標(biāo)準(zhǔn)物(10~180 kDa)評估樣品的分子質(zhì)量。
1.3.3 紫外-可見吸收光譜分析
參照文獻(xiàn)[23]方法,略作修改。將制備的凍干樣品用蒸餾水(pH 7.0)配制成0.25 mg/mL的溶液。測定波長294 nm和420 nm處的吸光度,A294nm和A420nm分別表示美拉德反應(yīng)中級階段產(chǎn)物和高級階段產(chǎn)物顏色深淺度的變化。采用蒸餾水作為空白參照。
1.3.4 內(nèi)源熒光光譜測定
采用F-7000熒光分光光度計(jì)測定樣品的熒光變化。用pH 7.0的10 mmol/L磷酸鹽緩沖溶液將樣品配制成質(zhì)量濃度為1 mg/mL。其中激發(fā)波長為295 nm,發(fā)射波長為300~400 nm,激發(fā)和發(fā)射狹縫均為2.5 nm,以未加樣品的緩沖液作為空白。
1.3.5 表面疏水性測定
參考文獻(xiàn)[24]的方法,采用ANS作熒光探針測定表面疏水性。稱取樣品溶于pH 7.0磷酸鹽緩沖溶液中,質(zhì)量濃度為1 mg/mL。取4 mL樣品溶液,加入20 μL ANS溶液,旋渦振蕩30 s后避光靜置10 min,使用F-7000熒光分光光度計(jì)測定,激發(fā)波長為390 nm,發(fā)射波長為400~600 nm,以熒光強(qiáng)度表示表面疏水性。
1.3.6 遠(yuǎn)紫外CD光譜分析
用磷酸鹽緩沖溶液配制成0.1 mg/mL蛋白樣品。采用光路長為0.1 cm的圓形石英比色皿進(jìn)行遠(yuǎn)紫外CD光譜分析,掃描范圍為185~250 nm。掃描9 次取平均值。
1.3.7 乳化性及乳化穩(wěn)定性測定
根據(jù)文獻(xiàn)[25]的方法,取6 mL樣品溶液,加入3 mL一級大豆油,使用ULTRA-TURRAX分散機(jī)以13 000 r/min均質(zhì)1 min,從底部吸取50 μL,立刻與5 mL 0.1% SDS混合搖勻。然后于500 nm波長處測吸光度A0。10 min后再次吸取50 μL乳狀液與5 mL 0.1% SDS溶液混合均勻,在500 nm波長處測吸光度A10,實(shí)驗(yàn)重復(fù)3 次取平均值。乳化性及乳化穩(wěn)定性按公式(1)、(2)計(jì)算:

式中:ρ為稀釋前的蛋白質(zhì)質(zhì)量濃度/(mg/mL);L為光程(1 cm);φ為形成乳液的油體積分?jǐn)?shù)(0.25%);DF為稀釋倍數(shù)(100)。
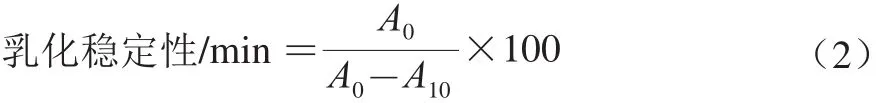
1.3.8 起泡性及泡沫穩(wěn)定性測定
參考文獻(xiàn)[26]的方法,取10 mL樣品于精密刻度管中,記錄最初體積V0,然后使用ULTRA-TURRAX分散機(jī)以13 000 r/min均質(zhì)1 min,記錄體積V1,靜置30 min后記錄體積V2。實(shí)驗(yàn)重復(fù)3 次取平均值。起泡性及泡沫穩(wěn)定性按公式(3)、(4)計(jì)算:
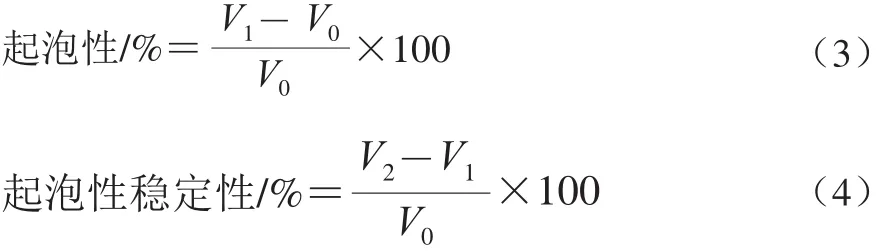
1.4 數(shù)據(jù)統(tǒng)計(jì)分析
實(shí)驗(yàn)結(jié)果重復(fù)3 次,采用Origin 8.0軟件繪圖,數(shù)據(jù)采用IBM SPSS Statistics 24軟件進(jìn)行單因素方差分析(ANOVA),P<0.05,差異顯著。
2 結(jié)果與分析
2.1 SDS-PAGE分析

圖1 不同添加順序形成的共價復(fù)合物和非共價復(fù)合物SDS-PAGE圖譜Fig. 1 SDS-PAGE profiles of covalent and non-covalent complexes formed by different addition sequences
SDS-PAGE主要用于表征β-LG與EGCG和Glc的共價結(jié)合過程。如圖1所示,天然β-LG分子質(zhì)量在18 kDa和36 kDa左右,這是因?yàn)樵谥行詐H值條件下β-LG的單體與二聚體共同存在。2 種不同添加順序的共價復(fù)合物的條帶都遷移較慢,復(fù)合物的分子質(zhì)量變大,表明EGCG、Glc與蛋白共價結(jié)合。此外,2 種共價復(fù)合物還出現(xiàn)了不同分子質(zhì)量的條帶,表明β-LG與EGCG、Glc之間還發(fā)生了蛋白質(zhì)交聯(lián)。Kroll等[27]研究發(fā)現(xiàn),EGCG在堿性條件下氧化成醌后,會更容易與蛋白質(zhì)側(cè)鏈的賴氨酸、色氨酸等親核基團(tuán)發(fā)生結(jié)合,同時作為一個親電子中間體更容易與蛋白質(zhì)形成交聯(lián)聚合物。而非共價結(jié)合β-LG-Glc-EGCG mix、β-LG-EGCG-Glc mix的2 種復(fù)合物與天然β-LG的條帶相似,沒有發(fā)生明顯的條帶遷移。主要是由于SDS-PAGE中,SDS和β-巰基乙醇容易破壞蛋白質(zhì)與多酚及糖之間的非共價作用健,而破壞不了共價作用鍵。
2.2 紫外-可見吸收光譜分析結(jié)果
美拉德反應(yīng)的程度可以通過反應(yīng)產(chǎn)物的顏色深淺反映,在波長294 nm和420 nm處的紫外-可見吸光度可以表示美拉德反應(yīng)產(chǎn)物的進(jìn)程。A294nm表示無色中間產(chǎn)物,A420nm表示末期褐變化合物[23]。由圖2可知,形成共價復(fù)合物后,A294nm和A420nm都明顯上升,這表明共價復(fù)合物產(chǎn)生了美拉德中間化合物,其中β-LG-Glc-EGCG con復(fù)合物在波長294 nm和波長420 nm處的吸光度都較β-LG-EGCG-Glc con復(fù)合物高。蛋白質(zhì)與多糖發(fā)生美拉德反應(yīng)時,蛋白質(zhì)的氨基與Glc的羰基結(jié)合[28]。而多酚也會與蛋白質(zhì)中的氨基酸殘基結(jié)合[29]。蛋白質(zhì)與Glc先反應(yīng)可以使更多的氨基與Glc的羰基結(jié)合,美拉德反應(yīng)產(chǎn)物也更高。這也表明不同的添加順序形成的共價復(fù)合物產(chǎn)生的美拉德反應(yīng)進(jìn)程不同。對于不同添加順序形成的非共價復(fù)合物的吸光度與對照組間并沒有明顯的差異(P>0.05),表明不同添加順序形成的2 種非共價復(fù)合物未發(fā)生美拉德反應(yīng),與SDS-PAGE結(jié)果一致。
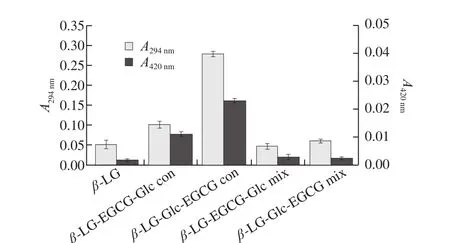
圖2 294 nm和420 nm波長處β-LG及其共價復(fù)合物和非共價復(fù)合物紫外-可見吸收光譜圖Fig. 2 UV-Vis absorption spectra at 294 nm and 420 nm
2.3 內(nèi)源熒光析

圖3 β-LG及其共價復(fù)合物和非共價復(fù)合物內(nèi)源熒光光譜圖Fig. 3 Intrinsic fluorescence spectra
β-LG中含有色氨酸和酪氨酸殘基,屬于內(nèi)源性熒光物質(zhì)。通過內(nèi)源熒光光譜可以分析β-LG的構(gòu)象變化。本實(shí)驗(yàn)采用激發(fā)波長295 nm,酪氨酸及苯丙氨酸不被激發(fā),可以認(rèn)為內(nèi)源熒光來自色氨酸殘基[30]。由圖3可知,2 種不同添加順序的非共價三元復(fù)合物的熒光強(qiáng)度降低,但β-LG-EGCG-Glc mix(1 538 nm)和β-LG-Glc-EGCG mix(1 481 nm)兩者相差不大,且沒有出現(xiàn)明顯的紅移現(xiàn)象。而共價三元復(fù)合物較非共價復(fù)合物有更顯著的熒光猝滅,熒光強(qiáng)度降低顯著。β-LG-EGCG-Glc con和β-LG-Glc-EGCG con的熒光強(qiáng)度分別為545.9 nm和271.3 nm。這可能是共價復(fù)合物一方面形成了緊密的結(jié)構(gòu)掩蓋了色氨酸殘基[31-32],另一方面可能色氨酸參與了共價反應(yīng)。劉夫國[33]在研究乳鐵蛋白與3 種多酚共價結(jié)合發(fā)現(xiàn)乳鐵蛋白的游離色氨酸含量減少,參與了共價反應(yīng)。此外,β-LG-Glc-EGCG con有明顯的紅移現(xiàn)象(8 nm),而β-LG-EGCG -Glc con沒有明顯的紅移,說明不同添加順序形成的共價復(fù)合物會使蛋白產(chǎn)生不同的構(gòu)象變化。Liu Fuguo等[21]研究綠原酸與乳鐵蛋白和Glc共價復(fù)合物發(fā)現(xiàn),共價復(fù)合物比非共價復(fù)合物具有更低的熒光強(qiáng)度。本實(shí)驗(yàn)結(jié)果與Liu Fuguo等[21]的研究結(jié)果一致。
2.4 表面疏水性結(jié)果

圖4 β-LG及其共價復(fù)合物和非共價復(fù)合物表面疏水性圖Fig. 4 Surface hydrophobicity spectra
蛋白質(zhì)表面疏水性的變化可以通過ANS法檢測[34]。有研究報道,ANS與β-LG有2 個結(jié)合位點(diǎn),一個作用在外部β-桶與α-螺旋間的疏水表面,一個作用在β-桶的內(nèi)部疏水區(qū)域[35-36]。由圖4可知,非共價復(fù)合物和共價復(fù)合物的熒光強(qiáng)度都增加,但不同添加順序的非共價復(fù)合物熒光強(qiáng)度沒有差異(561.3、577.5 nm)。而共價復(fù)合物相差顯著(1 024、4 616 nm),表明不同的添加順序形成的共價復(fù)合物在表面疏水性上有很大差異。共價復(fù)合物均出現(xiàn)明顯藍(lán)移(49、51 nm),表明蛋白質(zhì)的疏水基團(tuán)結(jié)構(gòu)暴露。可能的原因是EGCG和Glc與蛋白質(zhì)的親水基團(tuán)共價結(jié)合導(dǎo)致蛋白質(zhì)內(nèi)部結(jié)構(gòu)改變,疏水基團(tuán)被暴露。
2.5 CD光譜分析結(jié)果
遠(yuǎn)紫外CD光譜范圍為178~250 nm,可以反映蛋白質(zhì)等生物大分子主鏈的構(gòu)象信息。包括α-螺旋、β-折疊以及無規(guī)卷曲等信息[30]。β-乳球蛋白包含3 個轉(zhuǎn)角的α-螺旋、9 個β-折疊股[37],210~220 nm之間有負(fù)峰為β-折疊。從圖5可以看出,不同添加順序形成的共價復(fù)合物的二級結(jié)構(gòu)發(fā)生不同程度的改變。β-LG-EGCG-Glc con與β-LGGlc-EGCG con的β-折疊峰都發(fā)生了藍(lán)移,表明β-LG的β-折疊含量發(fā)生了改變。通過DichroWeb程序計(jì)算得到各復(fù)合物二級相對含量見表1。與對照蛋白相比,β-LG-EGCGGlc con的負(fù)橢圓率增大(β-折疊增至36.4%),而β-LGGlc-EGCG con的負(fù)橢圓率降低(β-折疊減少至32%)。同時由表1可知,這2 種共價復(fù)合物無規(guī)卷曲分別增加到34.6%和35.0%。Liu Fuguo等[21]發(fā)現(xiàn)乳鐵蛋白與綠原酸共價結(jié)合后再與葡聚糖共價結(jié)合,蛋白質(zhì)的α-螺旋減少,并且無序結(jié)構(gòu)增加。本實(shí)驗(yàn)中β-LG-EGCG-Glc con的二級含量變化趨勢與Liu Fuguo等[21]研究結(jié)果一致。不同順序形成的非共價復(fù)合物的結(jié)構(gòu)呈現(xiàn)相同的變化趨勢,β-LG-EGCG-Glc mix和β-LG-Glc-EGCG mix的β-折疊相對含量都增加,分別為36.2%和38.9%,無規(guī)卷曲相對含量則減少為27.8%和27.7%。

圖5 β-LG及其共價復(fù)合物和非共價復(fù)合物CD光譜分析Fig. 5 Circular dichroism (CD) spectra

表1 不同復(fù)合物的二級結(jié)構(gòu)相對含量Table 1 Contents of secondary structures of different complexes%
2.6 三元復(fù)合物的功能性質(zhì)測定結(jié)果
2.6.1 起泡性及起泡穩(wěn)定性

表2 不同復(fù)合物的功能性質(zhì)Table 2 Functional properties of different complexes
蛋白質(zhì)的擴(kuò)散能力、吸水及在空氣-水相界面展開重排的能力可以由起泡性能力表征[26]。由表2可知,共價復(fù)合物的起泡性比β-LG的起泡性大大提高,其中不同添加順序得到的共價復(fù)合物的起泡性差距明顯(P<0.05),β-LG-Glc-EGCG con的起泡性為121.87%,遠(yuǎn)高于β-LG-EGCG-Glc con(82.33%),不同添加順序?qū)ζ涔矁r復(fù)合物結(jié)構(gòu)的影響與其功能性質(zhì)的變化有關(guān)。而直接混合得到的不同添加順序的非共價復(fù)合物的起泡性雖較β-LG提高了10%左右,但非共價復(fù)合物兩者間的起泡性無明顯差別,熒光光譜和CD光譜結(jié)果表明不同添加順序得到的共價復(fù)合物結(jié)構(gòu)相差較大,而非共價復(fù)合物相差較小,起泡性的實(shí)驗(yàn)結(jié)果與之一致。此外,從起泡穩(wěn)定性的研究結(jié)果可知,β-LG-Glc-EGCG con的起泡穩(wěn)定性(64.57%)高于β-LG-EGCG-Glc con(54.20%),同時都遠(yuǎn)高于β-LG,而非共價復(fù)合物β-LGEGCG-Glc mix、β-LG-Glc-EGCG mix的起泡穩(wěn)定性分別為42.83%和48.73%。由此可知,EGCG和Glc及β-LG的復(fù)合物可以提高β-LG的起泡性及起泡穩(wěn)定性,其中,β-LGGlc-EGCG con復(fù)合物起泡性及起泡穩(wěn)定性效果最好,可能是因?yàn)棣?LG-Glc-EGCG con復(fù)合物分子間的作用力較強(qiáng),同時在溶液中的黏度最大,因此出現(xiàn)較好的穩(wěn)定性[32]。
2.6.2 乳化性及乳化穩(wěn)定性
乳化性的大小可以用作考量蛋白質(zhì)協(xié)助穩(wěn)定乳液能力的指標(biāo),而乳液維持其結(jié)構(gòu)的能力可以用乳化穩(wěn)定性來衡量[25]。如表2所示,復(fù)合物的乳化性均較β-LG有提高,其中,非共價處理得到的β-LG-EGCG-Glc mix、β-LG-Glc-EGCG mix乳化性分別為41.39%和41.86%,較高于共價處理得到的β-LG-EGCG-Glc con (32.45%)、β-LG-Glc-EGCG con(35.62%)。但從乳化穩(wěn)定性結(jié)果可知,共價復(fù)合物的乳化穩(wěn)定性高于非共價復(fù)合物。研究表明,蛋白質(zhì)的乳化性不僅與表面疏水性有關(guān),還與蛋白質(zhì)的溶解度及粒徑有關(guān)[24,38]。陳豪等[39]研究發(fā)現(xiàn)不同提取方法也會對乳化性有不同的影響。
3 結(jié) 論
本研究通過2 種方法制備了β-LG、EGCG、Glc的三元復(fù)合物,并討論了不同的添加順序?qū)?fù)合物結(jié)構(gòu)功能的影響以及共價與非共價復(fù)合物的區(qū)別。研究結(jié)果表明,不同添加順序?qū)矁r三元復(fù)合物影響較大,而對非共價復(fù)合物影響不明顯。4 種三元復(fù)合物的結(jié)構(gòu)發(fā)生了不同程度的改變,共價復(fù)合物較于非共價復(fù)合物有更大的結(jié)構(gòu)改變。其中,添加順序?qū)矁r復(fù)合物的結(jié)構(gòu)影響較大,β-LG-Glc-EGCG con比β-LG-EGCG-Glc con有更低的熒光猝滅以及更高的疏水性。而對于非共價復(fù)合物,不同的添加順序?qū)ζ溆绊懖幻黠@。乳化性和起泡性功能性質(zhì)結(jié)果表明共價復(fù)合物的功能性質(zhì)優(yōu)于非共價復(fù)合物,共價復(fù)合物中β-LG-Glc-EGCG con的功能性質(zhì)均優(yōu)于β-LG-EGCG-Glc con。而不同添加順序形成的非共價復(fù)合物沒有明顯差別。
