有機金屬鹵化物鈣鈦礦中的離子遷移現象及其研究進展*
2019-09-04 07:14:42王繼飛林東旭袁永波
物理學報 2019年15期
關鍵詞:研究
王繼飛 林東旭 袁永波?
1)(中南大學物理與電子學院,超微結構與超快過程研究所,長沙 410083)
2)(湖南理工學院物理與電子科學學院,岳陽 414006)
1 引 言
近年來,鹵化物鈣鈦礦材料以其較大的載流子擴散長度(單晶內大于100 μm)、容易調節的帶隙寬度[1,2]、高缺陷容忍度[3]、低制造成本以及較短的能量回收周期(0.22年)等優點[4],起了科研人員的廣泛關注.目前實驗室制備的鈣鈦礦太陽能電池的認證效率已達24.2%[5],展示出巨大的商業化前景.
盡管鹵化物鈣鈦礦太陽電池效率的提升非常迅速,但由于鈣鈦礦材料本身穩定性不佳,目前鈣鈦礦太陽能電池的實用化進程仍然面臨瓶頸.鈣鈦礦材料在濕氣作用下容易發生分解,加速器件老化.這種由于吸潮所導致的老化可通過對器件進行封裝而解決.然而,鈣鈦礦材料在高溫、光照或電場下的分解,則是當前仍需解決的問題[6?11].從材料自身的微納構成進行改進是解決以上問題的重要途經.例如,近年來的研究表明[12?14],將鈣鈦礦材料從三維(3D)降到準二維(quasi-2D)可顯著提高材料的濕度和熱穩定性.因此,進一步加深對鈣鈦礦材料物化性質的理解[15,16],不僅有利用于繼續提高鈣鈦礦電池效率,也有望為改善鈣鈦礦電池的穩定性指出方向.大量實驗和理論計算表明,存在離子遷移是鹵化物鈣鈦礦材料的重要特性之一.帶電離子的遷移和積聚直接導致鈣鈦礦薄膜的摻雜濃度以及內建電場的顯著變化,甚至可引起局部晶體結構的改變,是影響鈣鈦礦器件工作穩定性的關鍵因素[16?22].
本綜述以當前典型的鹵化物鈣鈦礦(如MAPbI3)為主要研究對象,首先介紹了離子遷移的研究背景,然后基于目前建立的理論解釋和實驗結果,對鹵化物鈣鈦礦材料中離子遷移的基本特點進行概括;討論了離子遷移對鈣鈦礦太陽能電池性能的影響;最后對抑制離子遷移方面的研究進展進行小結和展望.
2 鹵化物鈣鈦礦中的離子遷移
2.1 逐漸被重視的離子遷移
在鈣鈦礦太陽能電池的電流-電壓曲線(I-V曲線)測試中,研究人員經常可觀察到一種稱為“電流遲滯”的現象[21,23?27],即改變電壓掃描方向和速度時,多次測量獲得的電池I-V曲線不能重合,如圖1(a)所示[28].這一現象最早在2013年美國材料研究學會(MRS)秋季大會上被正式提出.McGehee團隊[22]和 Snaith團隊[20]同時報道了以介孔TiO2為電子傳輸層的鈣鈦礦太陽能電池中的電流遲滯現象.由于電流遲滯現象與測試時外加電壓的掃描方向和掃描速度等因素相關,如圖1(b)所示,因此電流遲滯的存在對電池性能的準確表征產生了顯著干擾.此外,電流遲滯現象的產生根源極有可能與制約電池長期工作穩定性的因素存在關聯[19,29?31],因此揭示電流遲滯現象的形成機理并對其進行有效抑制對于進一步提升鹵化物鈣鈦礦太陽能電池綜合性能至關重要.

圖1 (a)典型的平面型鈣鈦礦電池正反掃I-V曲線圖,掃描速度為0.20 V/s,I-V曲線正反掃不重合,表現出電流遲滯現象[28];(b)影響電流遲滯的因素和(c)導致電流遲滯的可能原因Fig.1.(a)Typical I-V character of the planner structure perovskite solar cell with the scan rate was 0.20 V/s,which expressived the obvious hysteresis;(b)the parameters lead to the hysteresis and(c)the possible reasons responsible for the hysteresis.
研究發現,電流遲滯現象不僅與器件測量的方式(如掃描速度、步長或方向)有關,還與器件結構以及器件掃描前的預處理方式(如光浸泡或外加偏壓)密切相關,如圖1(b)所示[19,32,33].目前普遍認為基于二氧化鈦(TiO2)的正置結構太陽能電池比基于富勒烯的倒置結構太陽能電池具有更明顯的電流遲滯現象[32].Snaith團隊[20]曾從原理上概括形成電流遲滯現象的三種可能原因,如圖1(c),即電荷陷阱效應(charge trap)、鐵電效應(ferroelectricity effect)和離子遷移(ion migration).到目前為止,理論計算和實驗結果表明鹵化物鈣鈦礦材料中的離子遷移是形成電流遲滯的重要貢獻之一[20,34?40].
研究人員對全無機鹵化物鈣鈦礦中的離子遷移很早就有研究.1983年,Fueki研究組[41]研究了無機鹵化物鈣鈦礦材料(CsPbCl3,CsPbBr3和KMnCl3)的離子導電特性,證實其離子導電性來自于鹵素空位的遷移.然而,以往關于無機鹵化物鈣鈦礦中離子遷移現象的研究一般是在200 ℃以上的高溫進行,遠遠高于實際太陽能電池的工作溫度.2014年,黃勁松團隊[42]率先證實有機-無機雜化鈣鈦礦材料在室溫下存在顯著的離子遷移行為.研究發現無載流子阻擋層的鈣鈦礦太陽能電池存在可翻轉的光伏效應(圖2(a)),其中光電流的變化幅度可以達到±20 mA/cm2(圖2(b)),遠大于光電流遲滯中的電流變化幅度(圖1(a)).基于可翻轉的光伏效應,黃勁松團隊進一步制作了基于甲基銨鉛碘(MAPbI3)鈣鈦礦的橫向結構器件(圖2(c)),并將兩電極之間的鈣鈦礦區域直接放置在顯微鏡下原位觀察,發現了持續外加電場(1.2 V/μm)的極化作用導致的薄膜形貌和吸光特性的變化.實驗結果表明,靠近陽極區域的薄膜因離子遷移導致材料分解而逐漸變得透明(圖2(d)),首次證實了雜化鹵化物鈣鈦礦中存在顯著的離子遷移現象[42].隨后的開爾文探針原子力顯微鏡(KPFM)測試發現,薄膜的表面勢在離子遷移前后發生了明顯變化(圖2(e)),結果表明離子的重新分布可以導致局部化學摻雜并影響薄膜內建電場的形成(圖2(f))[43].Zhang等[44]通過對MAPbI3納米線的研究,估算出室溫下MAPbI3鈣鈦礦中有 30% 的 MA+(離子濃度可達 1020cm–3)離子可以發生遷移.除實驗上觀察到離子遷移現象外,在理論上,研究者基于第一性原理計算也得到了鹵化物鈣鈦礦薄膜中不同種類離子發生遷移所需克服的能壘-活化能.Eames等[34]估算了鈣鈦礦材料內不同種類離子的遷移活化能,并給出了I–離子沿PbI6八面體邊緣移動時的弧線形軌跡(圖2(g)),其對應的離子遷移活化能為0.58 eV,低于其他可能的離子遷移路徑所需要克服的能壘.
2.2 遷移離子的種類
鹵化物鈣鈦礦的結構式為ABX3,其中A為有機陽離子(如MA+,FA+),B為金屬陽離子(Pb2+,Sn2+,Ge2+等),X為鹵素陰離子(I–,Br–,Cl–).通常情況下,尺寸較小的離子在遷移過程中引起的晶格形變小,因此更容易發生遷移.雖然I–離子半徑(206 pm)相對較大,但是無機鹵化物鈣鈦礦(CsPbI3和CsPbBr3)中的離子遷移現象(>200 ℃)已被證明是由I–空位遷移所主導[41].值得注意的是,I–離子存在于 MAPbI3鈣鈦礦的八面體的頂點,與最近鄰離子的距離約 4.46 ?.對比MA+和 Pb2+離子之間距離(6.28 ?),I–離子是MAPbI3鈣鈦礦中遷移路徑最短的離子[45].較短的離子遷移路徑有利于降低離子遷移活化能[34,40].
一般情況下,晶體內存在的缺陷為可動離子提供遷移的路徑.晶體內部的點缺陷包含Schottky缺陷(圖3(a))和 Frenkel缺陷(圖3(b)).其中,Schottky缺陷是晶格中同時存在、數量上滿足電中性條件的陰陽離子空位,通常在晶體形成的過程中產生或在晶體形成之后在靠近表面區域通過熱激發而產生,并擴散入晶體內部.對于MAPbI3鈣鈦礦,最容易形成的Schottky缺陷對是MA空位和 I空位(即和),如圖4(a)和圖4(c)所 示, 而Pb2+離子的空位缺陷()因存在較大形成能壘而較少形成.Frenkel缺陷是指在理想的晶體內部,由于熱激發產生空位的同時在附近晶格中產生相應的填隙離子形成成對的點缺陷.這些填隙離子和空位既可以熱激發成對產生,也可以相遇后發生湮滅,總體上使填隙離子和空位的濃度達到動態平衡[46].到目前為止,大量證據表明,在鈣鈦礦薄膜中可遷移的離子除了材料內部的組成成分離子(I–和 MA+)外[34,42,43,47],還有 MA 填隙離子(),I填隙離子(),MA 空位()和 I空位()[40,48].此外,還有制備過程中引入的氫離子雜質和來自電極處的金屬雜質等都可能參與離子遷移[36,49?51].圖4為MAPbI3鈣鈦礦材料中的組成成分離子、缺陷、填隙和雜質離子及其可能遷移的路徑示意圖,在沒有外加條件的影響下,這些可遷移離子的移動路徑具有隨機性.
另一方面,Pb2+離子則被證明很難遷移[34,35].鈣鈦礦材料中的Pb2+具有毒性,對鈣鈦礦太陽電池的應用將造成制約,通過其他金屬離子部分或全部取代 Pb2+,如 Sn2+,Mn2+或者 Ge2+等[52?61],對鈣鈦礦電池的商業化應用具有重要作用[62,63],但由于其具有更高的復合損失以及Sn2+容易被氧化為Sn4+,從而制約了器件性能,當前轉換效率在13%左右[59,62],與鉛基鈣鈦礦電池仍有較大的差距.此外,金屬離子如 Bi3+,Sb3+,Cu2+,亦可取代Pb2+形成鈣鈦礦結構[64,65].尋找新的可取代Pb2+離子的無毒金屬離子而又不損失器件效率,對于鈣鈦礦太陽能電池的發展以及抑制和消除其離子遷移特性都具有重要作用,是未來鈣鈦礦電池發展的重要方向之一.
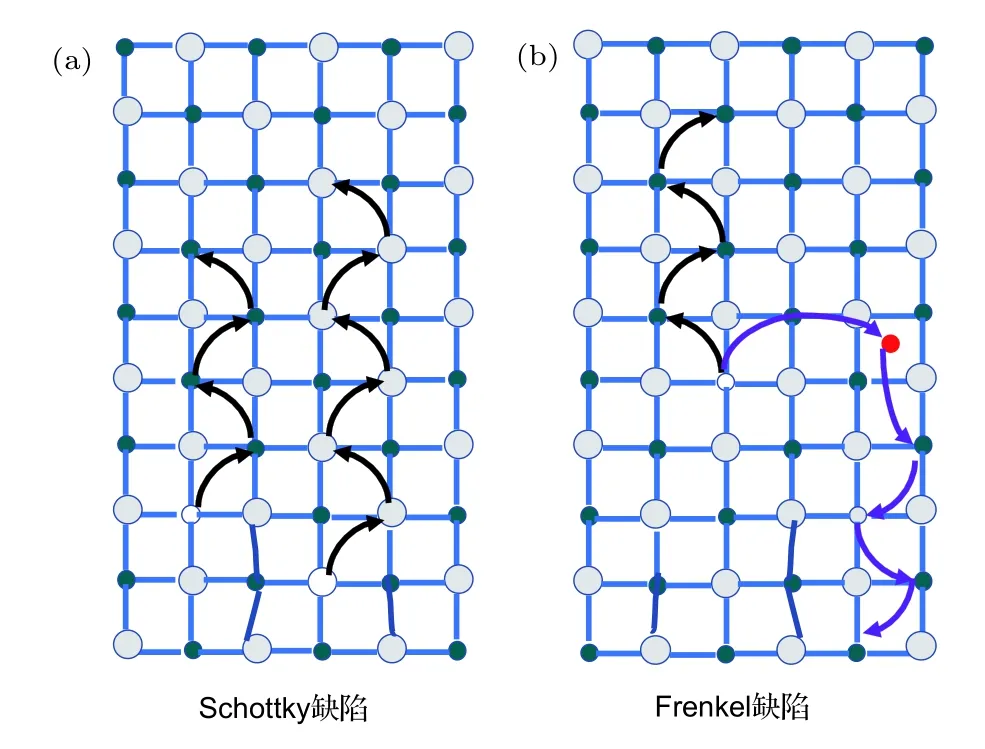
圖3 晶體中與離子移動相關的Schottky缺陷(a)和Frankel缺陷(b)的示意圖Fig.3.Illustrative diagrams of Schottky defects(a)and Frankel defect(b)related to ion migration in crystals.
由于離子遷移與缺陷的關系密切,而晶體中缺陷也會根據薄膜組分、制備工藝和后處理方法的不同而發生變化,因此可移動離子類型和濃度也隨之改變.例如,Cheng 等[66]研究提出,兩步旋涂法制備MAPbI3鈣鈦礦薄膜時,隨著MAI滴加的時間不同,薄膜內過量的 MA+離子濃度可在 1016cm–3到 1017cm–3范圍內變化.另外,Dhar等[67]指出,在由PbI2單晶轉換而來的MAPbI3單晶中同時存在著大量的(MA空位)和(I空位)離子.因此在兩步法制備的多晶MAPbI3薄膜中,占主導的可遷移離子一般是.此 外,熱 激 發Frenkel缺陷也會影響鈣鈦礦材料中的可移動離子濃度,Yang等[46]提出,由于空位周圍晶格的結構弛豫作用可降低空位與填隙離子間的湮滅概率,導致薄膜中可移動離子濃度逐漸上升.

圖4 MAPbI3鈣鈦礦材料中的可遷移離子、缺陷、填隙和雜質離子及其可能遷移的路徑示意圖,包括I空位(a),填隙(b),MA空位(c),填隙(d),以及I離子、MA離子和其他存在的雜質離子(e)Fig.4.Schematic illustration ofmobile ions,defects,interstitial and impurity ions in MAPbI3 and their possible migration paths,including,I vacancies(a),interstitialI ions(b),MA vacancies(c),interstitial MA ions(d),I ions,MA ions,and extrinsic impurities(e).
此外,由于鹵化物鈣鈦礦薄膜為多晶薄膜,晶界的存在為離子移動提供了廣泛的空間,對于離子遷移有重要影響.如Shao等[37]的研究證明,多晶鈣鈦礦薄膜中的晶界是產生離子遷移的主要區域.
2.3 離子遷移活化能EA
離子發生遷移的幾率(rm)取決于該離子在晶體中跳躍一次需要克服的能壘(即活化能,EA)和環境溫度,并存在如下關系:
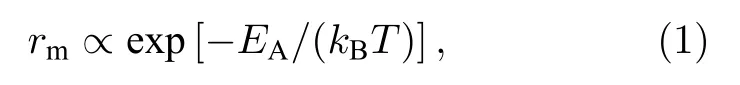
其中kBT是熱活化能,kB和T分別是玻爾茲曼常量和溫度.離子遷移活化能(EA)越小,離子的遷移越容易發生.
基于第一性原理計算,目前已有多個研究團隊模擬了在近鄰晶胞中存在空位(Schottky缺陷)的情況下,A位、B位和X位離子遷移的相應活化能[34,40,48,68].Eames 團隊[34]的計算結果表明,當 I–離子沿著八面體中的I-I邊界移動時(圖5(b)),離子移動具有最低的活化能 0.58 eV.當 MA+離子沿著單元晶胞移動時,由于面對著四個I–離子(圖5(c)),因此擁有更高的活化能 0.84 eV.當Pb2+離子沿著立方晶胞的對角線方向遷移時方向,圖5(b)),遷移能量勢壘最高為 2.31 eV.通過擬合瞬態光電流隨溫度和時間的變化關系,該團隊從實驗中獲得了I-離子移動活化能EA為0.60—0.68 eV,該結果與理論計算值吻合.因此,Eames 等[34]提出I–離子最容易移動,其擴散系數在320 K時估計為 10–12cm2·s–1.此外,Azpiroz 等[40]在同一時期也研究了鈣鈦礦材料中的各種缺陷對離子遷移的作用,提出了這些缺陷可能發生遷移的路徑,如圖5(d)—(g),并計算得出的活化能分別為 0.08,0.46 和0.80 eV,而離子根據遷移模式的不同其活化能為0.08 eV 或者 0.16 eV.關于缺陷對離子遷移的作用,Haruyama 團隊[48]計算得到了和的遷移活化能分別在 0.32—0.45 eV和 0.57—0.89 eV之間.總之,由于模型的不同,計算同種離子活化能數值并不完全一致,但是這些研究均表明,在MAPbI3中I–離子比其他離子更容易遷移.對于MAPbI3各種離子的遷移活化能總結在表1中.
2.4 離子遷移的實驗研究
實驗上,MA+離子在室溫下的移動首先被直接證明.由于 MA+離子在波數為 1468 cm-1的紅外區域有吸收峰,因此可利用光熱誘導共振(PTIR)顯微鏡技術原位檢測該離子在微米尺度的空間分布.在對 MAPbI3橫向器件電極化(1.6 V/μm)100—200 s后,如圖6(a),Yuan 等[43]原位觀察到MAPbI3薄膜陽極附近以及中間區域的MA+離子的減少和陰極附近MA+離子的聚集,直接證實了MA+離子在弱電場作用下的長程遷移.通過對MAPbI3薄膜進行變溫測量離子電導,Yuan等[43]利用阿倫尼烏斯方程擬合得出MA+離子遷移的活化能為0.36 eV.由實驗測量的MA+離子活化能小于由第一性原理計算所得結果(0.55 eV[48],或者0.84 eV[40];如表1 所列).導致活化能數值差異的可能的原因包括:理論計算得出的是晶體內部的離子遷移活化能,而實驗測量值可能主要由發生在晶體表面或晶界處的離子遷移所決定.此外,MA+離子的遷移能力顯著地受到水汽吸附的影響.Leijtens團隊[69]采用俄歇電子光譜法研究了外界環境條件對MAPbI3薄膜中離子遷移的影響:在環境濕度的影響下,MA+離子的遷移變得比I–離子的遷移更為顯著.
關于I–離子遷移的測量,Li團隊[70]利用空間分辨的X射線光電子譜(XPS)原位地證明了I–離子的遷移及其反向擴散行為.實驗中,Li等[70]通過對橫向結構(圖6(b))的MAPbI3–xClx器件進行電場極化處理,同時利用光斑直徑為50 μm的XPS技術研究了薄膜中不同位置的I/Pb比例及其變化.實驗發現陽極附近的I/Pb比例在電場極化處理后由3.03提高到5.65,與此同時陰極附近的I/Pb比例降低至小于3,證實了外加電場下I–離子的遷移,如圖6(c).此外,在撤銷極化電場的6 h后,樣品中不同位置的I/Pb比例恢復到3,表明室溫下聚集的I–離子存在明顯的反向擴散現象.另一方面,更多的實驗證據側面證明室溫下I–離子可以發生遷移.例如,de Bastiani團隊[71]發現在用銀作為MAPbI3橫向器件的電極時,當器件在電場極化下,陽極電極發生損壞,該損壞是由于銀電極與來自MAPbI3中移動的I–離子發生化學反應生成碘化銀所造成的.此外,Zhang團隊[72]研究發現,當逐漸提高基于MAPbI3–xBrx混合鹵素發光器件的驅動電壓時鈣鈦礦的電致發光光譜出現藍移,出現這一現象的原因可能與發光區中I–離子數量的減少有關.
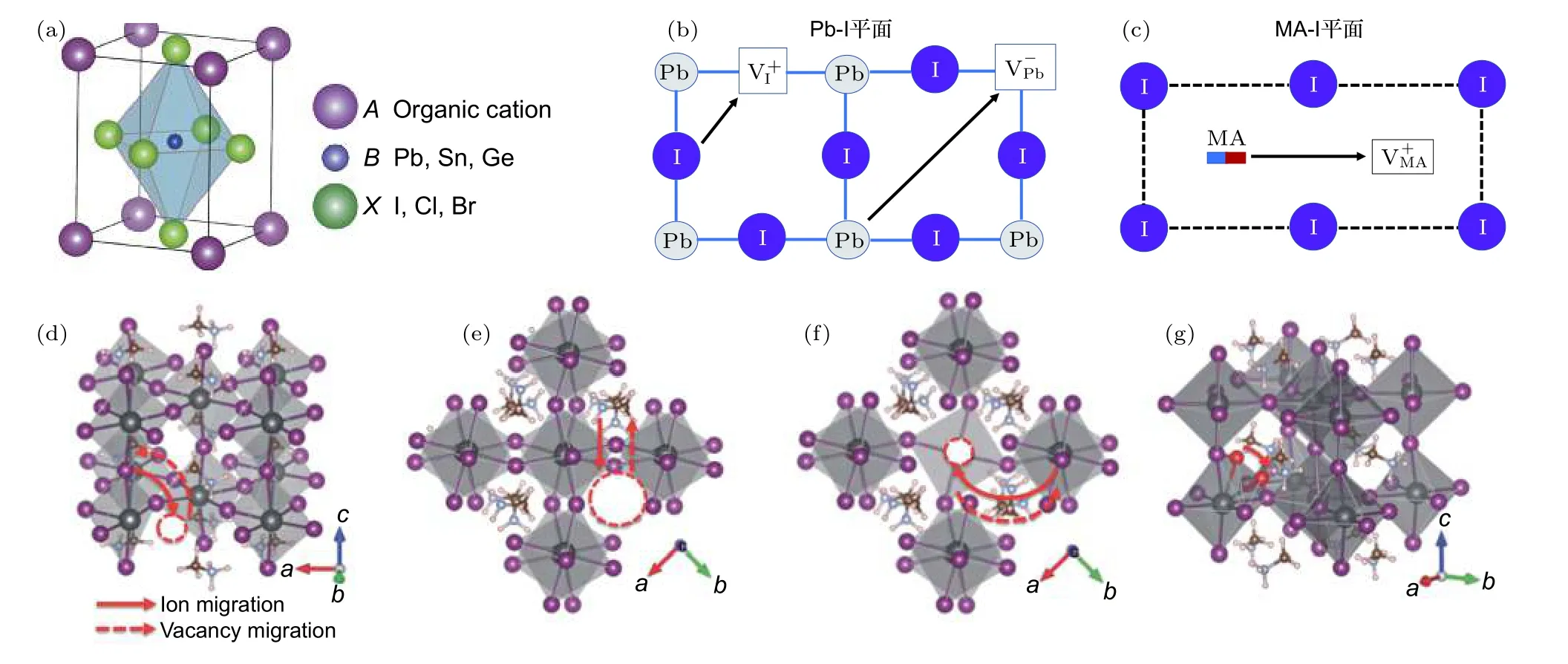
圖5 (a)鈣鈦礦晶體結構圖;(b)鈣鈦礦中 Pb-I平面中 I–離子移動路徑和(c)MA-I平面中 MA+離子移動的路徑[34];各種缺陷遷移路徑(d)和碘填隙遷移路徑(g)II[40]Fig.5.(a)Structure diagram of perovskite crystal;migration paths for I– ion in Pb-I plane(b)and MA+ ion in MA-I plane(c)[34];diffusion pathsfor(f)and interstitial iodine ion II(g)respectively[40].
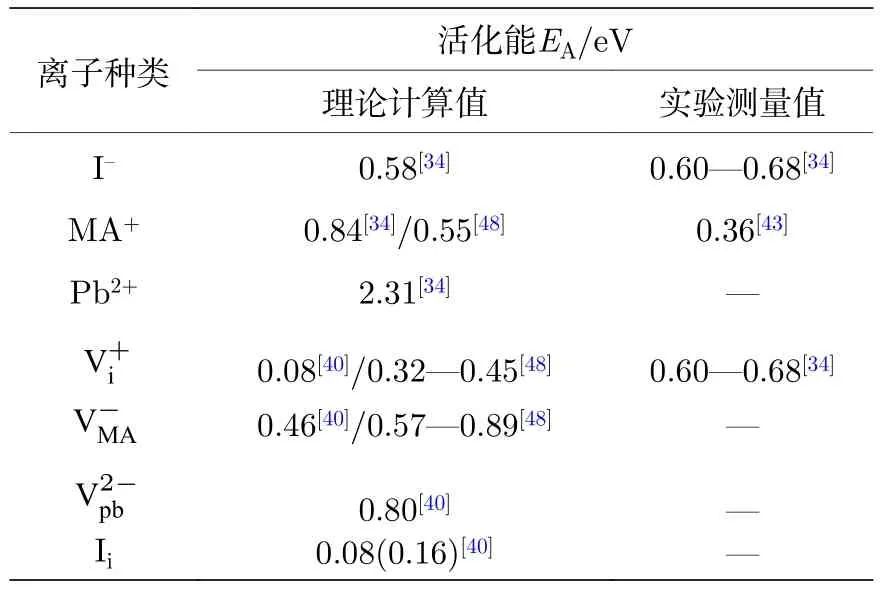
表1 理論計算和實驗測試的 MAPbI3 中可遷移離子的離子活化能對比Table 1. Comparison of ion activation energies(EA)between theoretical calculations and experimental measurements of the migration ions in the MAPbI3 film.
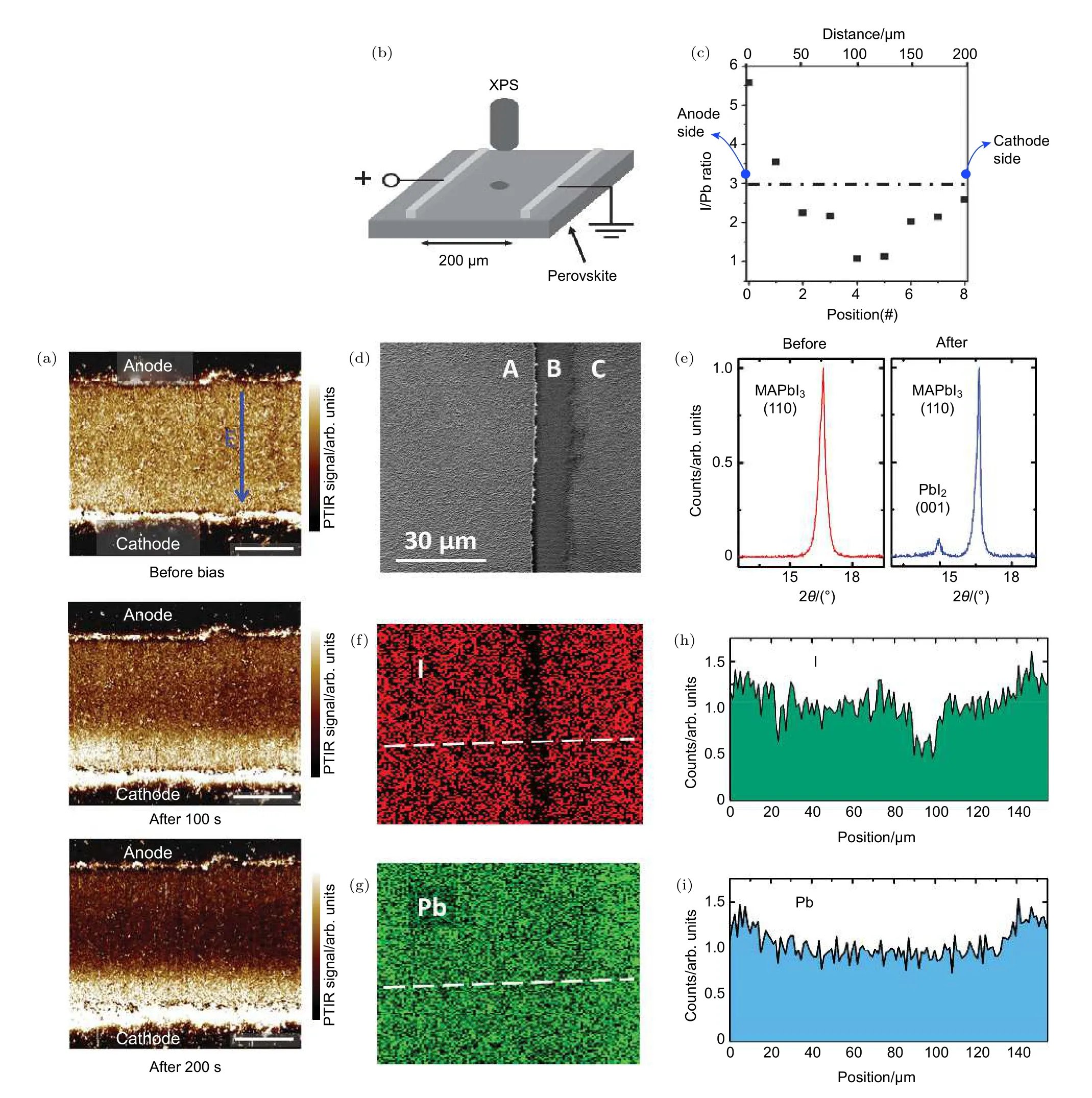
圖6 (a)PTIR 顯微鏡下 MAPbI3 薄膜在 1.6 V/μm 電場極化 100 和 200 s 前后,MA+離子的分布圖,其中電極間距為 100 μm[43];(b)實驗中用XPS方法于測試元素分布的橫向器件和實驗裝置示意圖及測得的從陽極到陰極之間的I/Pb比例分布圖(c),施加的直流電壓為 1 V,持續時間 30 min,兩電極間的橫向距離為 200 μm,其中位置“0”為接近正極處,位置“8”為接近陰極處[70];(d)橫向器件中具有PbI2條形區域的SEM和XRD分析,對橫向結構的MAPbI3薄膜電場極化后具有典型的PbI2條形區域的SEM圖(d)和極化前后的XRD圖(e);(f),(h)EDX測試下的具有PbI2條形區域的橫向鈣鈦礦器件的碘元素(f)和鉛元素(h)分布圖;(g),(i)分別為碘元素和鉛元素在(f),(h)白線處的濃度變化曲線[35]Fig.6.(a)Corresponding PTIR images for the CH3 asymmetric deformation absorption of the methylammonium ion(1468 cm–1)obtained before,after 100 s and after 200 s electrical poling,respectively.The poling field was 1.6 V/μm and the distance between the electrode is 100 μm[43].(b)Illustration of the set-up for characterization of elements distribution using a lateral structure with XPS method and the obtained I/Pb ration against position from anode side to cathode side(c).The applied DC voltage is 1 V for 30 min.The lateral distance between two electrodes is 200 μm.Position 0 is near the anode and position 8 is near cathode side[70].(d)SEM and XRD study of the lateral devices with a PbI2 thread.By electric polarization of MAPbI3 films with lateral symmetric electrode structure,SEM image(d)of a typical PbI2 thread,(e)XRD spectra of the MAPbI3 film before and after the formation of a PbI2 thread.(f),(h)The distribution diagram of iodine(f)and lead element(h)of lateral perovskite device with PbI2 thread line under energy dispersion X-ray(EDX)test.(g),(i)The concentration curves of iodine(g)and lead(i)at the white line in Figs.(f)and(h),respectively[35].
如上所述,離子的遷移能力與溫度密切相關.在適當加熱(330 K)的情況下,Yuan 等[35]發現MA+離子和I–離子的遷移變得非常劇烈.實驗中觀察到劇烈的MA+離子和I–離子遷移可導致MAPbI3材料中出現可移動的條形區域,如圖6(d)中B區域.XRD測試表明可移動區域主要由PbI2相構成,如圖6(e).PbI2條形區域在外加電場(3 V/μm)下的移動通過光學顯微鏡可以直接觀察到.能量色散X射線(EDX)元素分析證實了大量 I–離子發生遷移(圖6(f)、圖6(g))的同時Pb2+離子不存在明顯遷移,如圖6(h)和圖6(i),這與Eames的理論計算得出Pb2+離子移動活化能高達 2.31 eV的結果[34]相一致,即 Pb2+離子在鈣鈦礦材料中難以遷移,或者只有在鈣鈦礦薄膜發生明顯分解時Pb2+才發生遷移.
3 離子遷移的影響
3.1 離子遷移對鈣鈦礦材料電子結構的影響
鈣鈦礦薄膜中的離子遷移可以導致點缺陷或雜質的聚集.這些缺陷通常情況下在鈣鈦礦晶體內部形成為淺能級缺陷,改變薄膜的電學性質.Yin[51]通過密度泛函理論(DFT)計算了在不同前驅體成分比例條件下(如圖7(b)中A為富I,B為I-Pb基本平衡,C為富Pb條件),生成的鈣鈦礦材料中缺陷能級的形成能,如圖7(c)—(e)和表2所示.計算結果表明,雖然點缺陷的形成能在不同前驅體成分比例條件下差別很大,但是通常容易形成的點缺陷為淺能級缺陷
值得注意的是,點缺陷仍有可能在晶體表面形成深能級缺陷,從而對薄膜電學性質產生影響.Xu等[73]通過DFT計算,得出鈣鈦礦表面過量的I–離子會導致Pb-I反位缺陷的形成.Pb-I反位缺陷可在導帶之下形成深能級陷阱.這些深能級陷阱的存在,將會成為非輻射復合中心,危害器件的性能.此外,點陷阱的能級深度與鈣鈦礦的組分相關.例如在FAPbI3中,IFA在I-rich的情況下形成能較低并且可在帶隙中形成深能級陷阱(0.2 eV[74].Zhao等[75]通過DFT計算發現離子遷移會誘導形成I三聚體和Pb二聚體,從而形成缺陷,其陷阱深度為 0.3 eV.
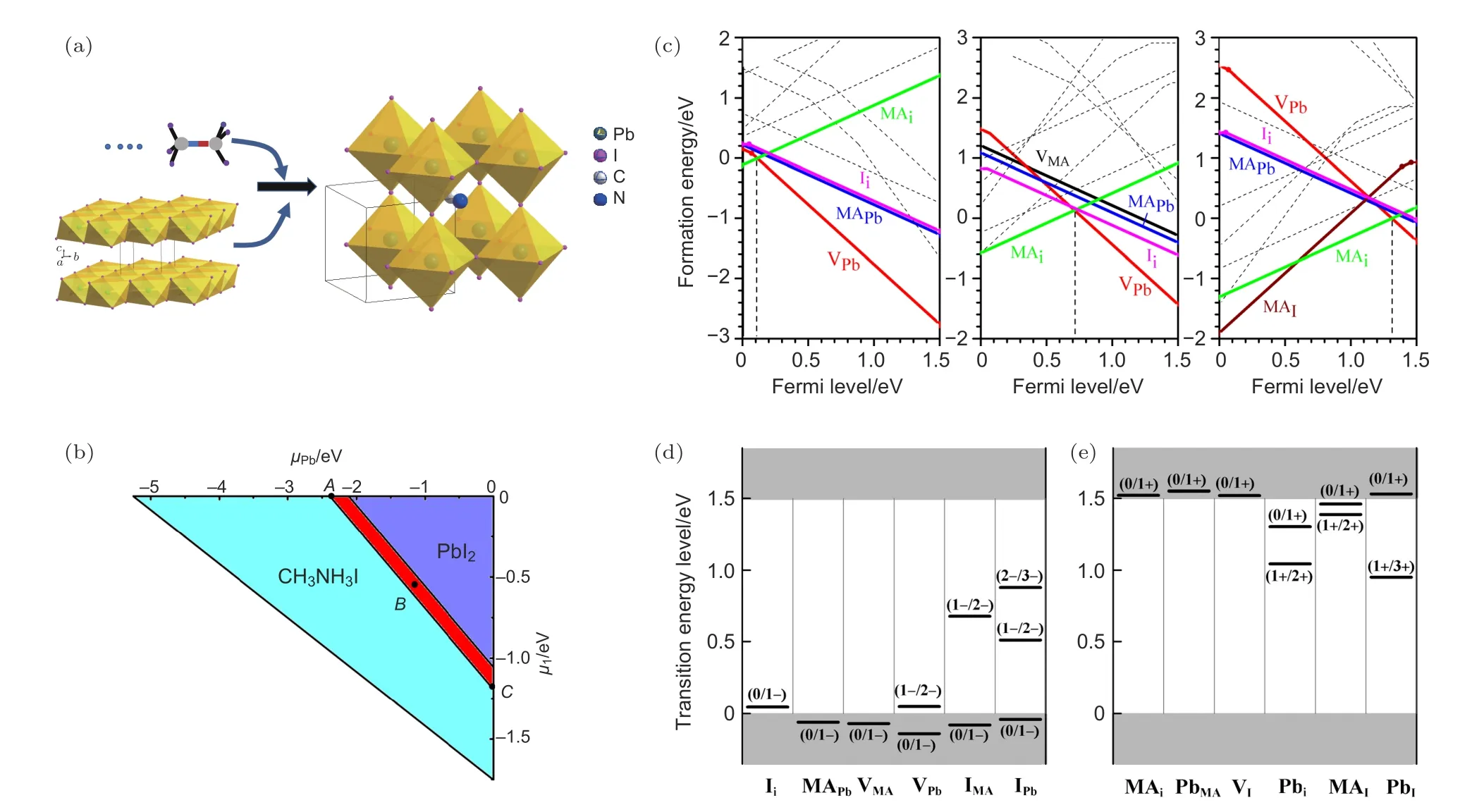
圖7 (a)MAPbI3 合成簡圖;(b)紅色區域為 MAPbI3 中達到熱力學平衡生長時的熱力學穩定區域;(c)MAPbI3 中化學勢在 A,B,C(圖7(b)中 A,B,C 處)位置時內部點缺陷的形成能,虛線代表具有更高形成能的缺陷;(d),(e)分別為通過計算得到的MAPbI3鈣鈦礦中內部電子受體和給體的傳輸能級[51]Fig.7.(a)Schematic diagram of the MAPbI3 unit cell.(b)The thermodynamic stability range for equilibriμm growth of MAPbI3 is in the red region.The formation energies of intrinsic point defects in MAPbI3 at chemical potentials A,B,and C shown in(c),where defects with much high formation energies have displayed as dashed lines.The calculated transition energy levels of(d)intrinsic acceptors and(e)intrinsic donors in MAPbI3.Dashed lines stand for the defects that have much higher formation energy so that they cannot exist[51].
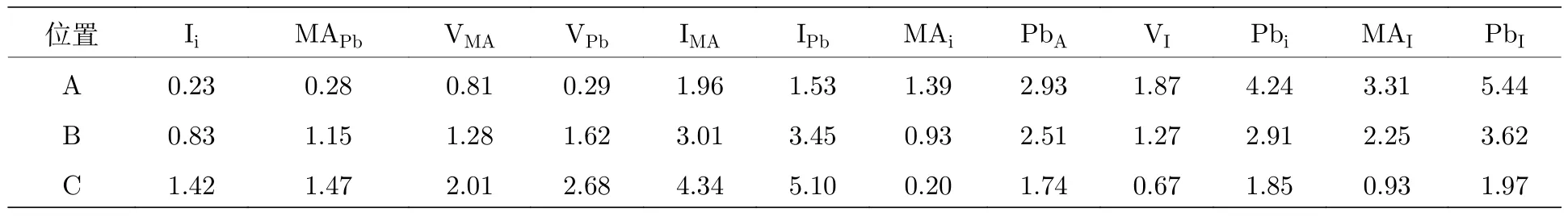
表2 在化學勢分別為 A,B,C,如圖7(c)中 A,B,C 處時,MAPbI3 鈣鈦礦材料中內部中性點缺陷的形成能(單位:eV)[51]Table 2. The formation energies(unit:eV)of the neutral defects in MAPbI3 at chemical potential points A,B,and C[51].
3.2 離子遷移對載流子傳輸與復合的影響
在外加電場的影響下離子將朝著特定的方向發生遷移,并在特定區域聚集.這些帶電離子的聚集會引起局部化學摻雜效應,改變離子聚集區域費米能級相對于導帶/價帶(VB/CB)的位置,使得能級發生彎曲,從而影響鈣鈦礦材料中光生載流子的拆分、傳輸與提取以及器件的性能.其中,正電荷缺陷的遷移會在鈣鈦礦的導帶附近形成較淺的能級態,從而導致鈣鈦礦與陽極接觸位置產生n型摻雜.反過來,負電荷離子的遷移可能會在價帶以上產生電子能級,從而導致鈣鈦礦與陰極接觸位置產生p型摻雜.Xiao等[42]報道的可翻轉光伏效應現象中,在正負外加偏壓極化下,鈣鈦礦材料中的離子遷移會形成p-i-n或者n-i-p兩種結構,從而出現了測試過程中電流方向完全相反的現象.此外,除了離子的化學摻雜效應,Eames等[34]從內建電場屏蔽的角度也進行了解釋.離子和帶電缺陷在內電場下發生定向運動,并在某些區域聚集,同時形成一個局部電場,如圖8(a),該電場與內電場方向相反從而對內電場部分屏蔽.在I-V測試過程中,這些離子和帶電缺陷在外加電場下再次發生定向運動,如正向偏壓下在界面處擴散而消失,產生的屏蔽電場消失,有利于光生載流子的傳輸與收集.相反,在反向偏壓時,離子與帶電缺陷的進一步遷移,提高了屏蔽電場,導致光生載流子的傳輸與收集更加困難,如圖8(b).因此,離子和帶電缺陷在外加電場下的定向運動對光生載流子的傳輸產生影響并形成遲滯現象.
離子遷移對于鈣鈦礦材料中載流子復合的影響,可通過觀察光致發光(PL)的強度的變化來直觀地進行研究.大量的觀測結果表明,由于MA+離子或者I–離子離子遷移而形成的離子積累可以使得積累區發生顯著的PL淬滅[42,47,66,76?78].相關的PL淬滅存在兩種作用機制:1)首先由于離子的化學摻雜效應,可在聚集區域形成高濃度的多數載流子(電子或者空穴),高濃度的多數載流子將顯著降低由光照產生的非平衡少數載流子的平均壽命,產生大量無輻射躍遷形成淬滅,對于MAPbI3多晶薄膜鈣鈦礦,研究表明,其離子聚集濃度可以達到1017cm–3甚至更高[44,79?85];2)如上所述,聚集在材料界面的離子有可能在鈣鈦礦晶體表面形成深能級陷阱態(如鈣鈦礦表面過量的I–離子導致的Pb-I反位缺陷),從而構成非輻射復合的中心,促使PL淬滅的增強.
雖然通常情況下離子遷移將導致PL強度的顯著下降.然而Hoke等[86]也報道過由于離子遷移導致的PL強度增強的情況.他們發現離子遷移會使具有混合鹵素的鈣鈦礦(MAPbBrxI3–x)中的鹵素離子在空間上的分布發生嚴重相分離,即光照后形成了具有不同晶格常數的富碘區和富溴區兩種鈣鈦礦相.這些結構具有不同的能級,其中富碘區的結構具有更小的帶寬而成為能量勢阱,這種能量勢阱同時對電子和空穴具有束縛作用,因此通過增強載流子相遇概率提高了PL強度,同時由于富碘區禁帶寬度變小,伴隨PL增強同時發生的現象是PL的峰值發生紅移.此外,Zhang等[44]在研究中發現,當MA+離子在陰極附近聚集時PL強度減弱,但是當 I–離子在陽極附近聚集時,PL 強度卻出現了逐漸增強的現象,這與通常觀察到的離子聚集誘導 PL淬滅現象不一致.隨后,Hentz等[87]在研究中也發現了I–離子聚集時PL強度增強的現象.關于PL強度在離子遷移時出現不一致的原因尚不清楚,可能是由陷阱的深度和摻雜濃度所決定.
應當指出,離子遷移對于PL的影響是可逆的.比如在MAPbBrxI3–x材料中,由于光照誘導相分離可產生PL強度和峰位變化,但是當把鈣鈦礦材料放置在暗態下幾分鐘后又會恢復到材料的初始狀態[86].同樣,在外加偏壓對材料進行極化過程中,在撤去外加偏壓使材料靜置一段時間后,由于離子的反向擴散效應,由離子聚集所表現出來的PL強度的變化也可以逐漸恢復.這種可逆有時候需要一定的外在輔助條件,如給材料一定的熱退火會有助于更好地恢復到材料的初始狀態.
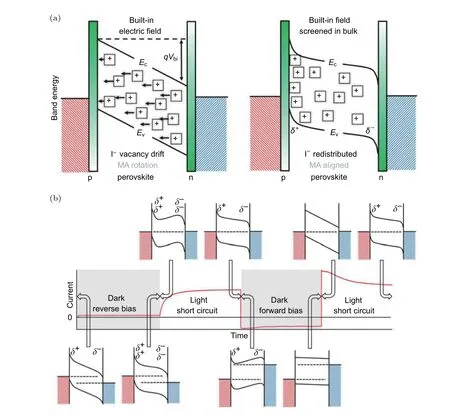
圖8 (a)短路時離子和帶電缺陷遷移對 p-i-n 器件能帶的影響,Ec 為導帶,Ev 為價帶,Vbi為內建電場;帶有加號的方格表示碘離子空位;(b)不同偏壓條件和時間下計時光電流測量時假設的能級結構圖,導帶和價帶的變化對應于碘離子空位在不同外加偏壓和作用時間下與界面之間的再分布[34]Fig.8.(a)Schematic representation indicating the impact of ion vacancy drift on the band energies of a p-i-n device at short circuit conditions.Ec is the conduction band energy,Ev is the valence band energy and Vbi is the built-in potential.The squares with“plus” signs indicate the iodide ion vacancies.Implicit in the diagram is that the vacancies with effective positive charges are balanced by immobile cation vacancies(not shown)with effective negative charges.(b)Hypothesized energy level configurations corresponding to different bias conditions and times during the chrono-photoamperometry measurements.The variation in the conduction and valence bands corresponds to the redistribution of iodide ion vacancies to and from interfaces with different applied potentials and times[34].
4 離子遷移的測試方法
如上所述,鹵化物鈣鈦礦材料中離子的發生遷移的幾率可由離子的遷移活化能(EA)衡量.根據(1)式,由離子遷移形成的電導率si在半對數坐標中與1/T存在線性關系.此外,考慮到熱激發作用下理想晶格中也能形成可移動離子,其形成的離子濃度通常具有如下關系(忽略可移動Pb離子的產生):
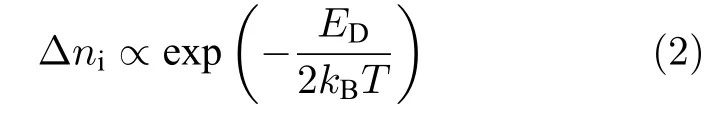
其中ED為產生可移動離子的缺陷形成能.因此通過(1)和(2)式,可知鈣鈦礦材料中的離子電導si為
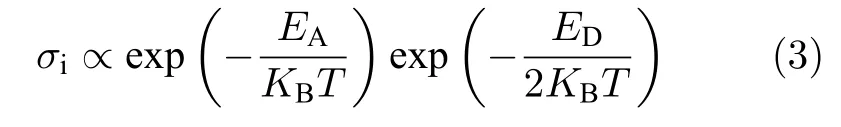
因此,用 l nσi-1/T的圖中的斜率可表述離子遷移的活化能EA(熱激發缺陷可忽略區間)或者活化能與形成能半值之和:(熱激發缺陷不可忽略區間).而這兩個區間的轉變點受薄膜中的初始非本征離子濃度(extrinsic defects)所影響,如圖9(a)所示.
某些特殊性情況下,由于晶體相變或者多種離子遷移通道共存,此時,l nσi-1/T的函數圖形將變得更加復雜.對于具有多種可遷移離子的材料,每一種可遷移離子具有各自的遷移活化能(EA),該值取決于離子發生一次跳躍時所面臨的能量勢壘,受晶體結構、離子半徑和離子價態的影響.一般來說,在材料中占主導作用的可遷移離子往往具有最低遷移活化能.
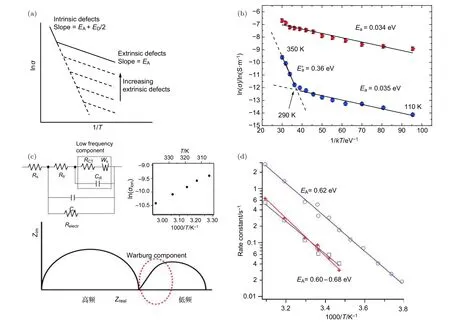
圖9 (a)固態薄膜中不同數量外部缺陷情況下典型的離子電導 l nσi-1/T 曲線圖,其中EA是離子遷移的活化能,ED是熱激發點缺陷的形成能;(b)通過變溫測試MAPbI3鈣鈦礦離子電導,結合阿倫尼伍茲方程擬合離子遷移活化能EA[43];(c)具有離子遷移過程的鹵化物鈣鈦礦的電化學阻抗譜圖,在低頻區域阻抗表現出線性變化,說明具有離子傳輸特性,其等效電路如圖中插圖,虛線框中為低頻區等效電路圖,圖中的Ws是Warburg阻抗單元,Rct是界面電荷傳輸阻抗,Cdl是所有在界面聚集的離子相關的德拜層電容[88];(d)通過瞬態光電流弛豫時間常數結合阿倫尼烏斯方程擬合離子遷移激活能[34]Fig.9.(a)Typical l nσi-1/T curves for ion conduction in solid with different amount of extrinsic defects,where EA is the activation energy for ion migration,ED is the formation energy for thermal-excited point defects.(b)Arrhenius plot of the conductivity of the MAPbI3 film under dark(blue)and illμmination(red,the light intensity is 0.25 mW/cm)[43].(c)Typical Nyquist plot for mixed conductor system with Warburg diffusion as evidence by the linear portion of the low frequency regime.Equivalent circuit diagram was showedin the figure and the dashed line islowfrequency ionic elements.Ws is the Warburg component,Rct is the interfacial charge transfer and Cdl is modeled as an interfacial an interfacial Debye-layer capacitance[88].(d)Calculation of activation energy of ion migration by transient photocurrent combined with Arius drawing method[34].
實驗上,目前測量離子遷移活化能的方法通常都是基于阿倫尼烏斯方程擬合,包括利用對溫度敏感的離子電導(圖9(a)、圖9(b))、Warburg時間常數(圖9(c))[88]和瞬態光電流弛豫時間常數(圖9(d))[34].圖9(b)為Yuan等[43]用低溫探針臺測試的橫向結構Au/MAPbI3/Au的離子電導,圖中紅色和藍色分別為光照和暗態條件下薄膜總電導,通過比較可知暗態下290—350 K范圍的電導主要由離子遷移貢獻,對其斜率擬合得出EA=0.36 eV.圖9(c)[88]為具有離子遷移特性的鹵化物鈣鈦礦的電化學阻抗譜,在低頻區存在一段線性的阻抗譜,表明存在一種具有擴散特征的阻抗單元(即Warburg component),通過對阻抗譜按照圖9(c)中等效電路進行擬合可獲得Warburg時間常數,再通過Warburg時間常數對溫度的依賴特性擬合可得離子遷移活化能EA.圖9(d)[34]為通過測試瞬態光電流弛豫過程來分析離子遷移活化能的阿倫尼烏斯擬合.具有離子遷移現象的鈣鈦礦電池其光電流從產生到飽和具有一個弛豫過程,通過指數擬合光電流達到飽和的過程可提取出弛豫時間常數,其弛豫時間常數與不同溫度下離子的移動速度相關,通過阿倫尼烏斯擬合也可獲得離子遷移活化能EA.
另一方面,移動的離子也可以從Tubandt方法[41]中推斷出來:離子導體被包含在電化學電池中,并對其進行長時間的直流偏置電壓極化(一般在數天左右).極化后,可以通過測量電化學電池中不同部分的重量和成分來分析由于離子移動產生的界面反應生成物.
5 離子遷移的抑制
大量的研究表明,離子遷移會導致鈣鈦礦薄膜相的不穩定,甚至造成薄膜的分解和器件的失效[42,89?92].提高鈣鈦礦太陽能電池的本征穩定性是實現提升相應電池壽命的重要途徑.目前,不少課題組在離子遷移對器件長期穩定性的影響方面也開展了相關研究[69,89,90,93?101].
研究表明,由光照[86,102]或外電場[35,103]引起的過量離子堆積及其所導致的鈣鈦礦晶體結構變化以及光電性能變化可在一定程度上恢復.例如暗態存儲[86,104]時薄膜中的可移動離子濃度可通過反向擴散或者離子-空位湮滅過程降低,而適當加熱處理可以使恢復過程加快[19,35,44].Hoke等[86]發現光照可導致MAPbBrxI3–x中出現相分離并產生新材料相的XRD衍射峰,但是在暗態下放置幾分鐘后薄膜的XRD衍射譜會逐漸恢復到初始狀態,如圖10(a),變化前后材料屬于具有不同晶格常數的鈣鈦礦相.此外,Yuan等[35]發現離子的移動還能導致鈣鈦礦與PbI2相之間的可逆轉變.研究中表明在外加電場的驅動下,MAPbI3中大量可移動離子(MA+,I–)被抽出后將變為具有層狀結構的PbI2晶體,如圖10(b).然而在 330 K 下,具有足夠活性的I–和MA+離子可以重新注入到PbI2晶體中將其轉變為 MAPbI3鈣鈦礦,從而實現MAPbI3晶體和PbI2晶體之間的可逆轉變.Yang等[105]在 323 K 下對(陽極)/MAPbI3/AgI/Ag(陰極)結構電池施加一周的直流偏壓,觀察到在(陽極)/MAPbI3界面處形成PbI2,這是由于I–離子移動到陽極與Pb發生反應生成了PbI2.在另一項研究中,Jeangros等[106]使用EDX測量I/Pb比率,發現偏置前后I/Pb比率由2.7降至2.4,這表明樣品中可能有一些I–離子隨著有機物一起揮發.de Bastiani等[71]對以 Ag為電極的橫向結構MAPbI3器件施加偏壓,觀察到只有正偏壓電極對應的區域被破壞,如圖10(c),這應該是由 I–離子向正偏壓電極遷移及其與Ag的反應引起的.Akbulatov等[107]通過飛行時間二次離子質譜法(TOF-SIMS)分析表明,MAPbI3分解產生的揮發性氣體通過富勒烯及其衍生物([6,6]-phenyl-C61-butyric acid methyl ester,PCBM)層,與 Ag 電極發生反應并形成界面AgI層.為了解決離子遷移引起的對功能層的腐蝕問題[108?110],研究者通過引入腐蝕阻擋層使活性層與金屬電極之間分離開[111?113].
抑制離子遷移是提高器件穩定性的的關鍵,目前已有不少課題組從抑制離子遷移的角度開展了一些相關研究[31,108,114,115].
5.1 界面鈍化
大量研究表明,晶界的存在為離子遷移提供了場所,是產生離子遷移的主要通道[37,116?118].關于減少晶界缺陷,首先一個途徑是提高晶粒尺寸,減少晶界面積.研究發現,具有大晶粒尺寸和較小晶界區域的鈣鈦礦器件通常具有可忽略的電流遲滯現象[37,79,116].Hu等[79]研究表明減少晶界面積對提高鹵化物鈣鈦礦(MAPbI3–xBrx,x<1)光穩定性具有重要作用.他們將MAPbI3–xBrx膜的晶粒尺寸從 200 nm增加到 1 μm以上,并進行長時間的光浸泡,發現具有大晶粒尺寸的 MAPbI3–xBrx膜的XRD峰沒有發生分裂.在另一項研究中,Xing等[119]分別對尺寸約為300 nm的小晶粒和約1 μm的大晶粒多晶鈣鈦礦薄膜進行溫度依賴性電導率測試,離子遷移活化能分別為0.27 eV和0.50 eV,如圖11(a),證實增加晶粒尺寸有利于抑制離子遷移.因此,通過制取晶界少、晶粒大的高質量薄膜可以有效地抑制離子遷移,提高薄膜穩定性[118,120,121].
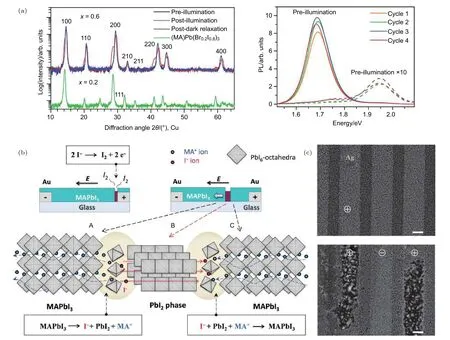
圖10 (a)MAPb(Br0.6I0.4)3的 XRD 和 PL 圖,左圖表示 MAPb(Br0.6I0.4)3膜在光浸泡前(黑色)和在約 50 mW·cm–2下白光浸泡5 min(紅色)以及置于暗態下 2 h(藍色)的 XRD 圖譜,右圖通過打開(457 nm,15 mW·cm–2)和關閉激發光(黑暗中 5 min 后),光譜可以在這兩種狀態之間重復循環[86];(b)電場下MAPbI3和PbI2之間的可逆轉換,左邊為橫向MAPbI3鈣鈦礦太陽能電池的橫截面圖,其中在陽極下方形成PbI2線,右圖具有PbI2線移向陰極的側向MAPbI3器件的圖示,這是由“區域A/區域B”和“區域B/區域C”界面上分別發生的兩組固態化學反應造成[35];(c)極化前(左)和極化后(右)帶銀電極的鈣鈦礦橫向結構的SEM俯視圖;“+”和“-”符號表示不同的電極極化[71]Fig.10.(a)XRD and PL of MAPb(Br0.6I0.4)3.The black line in the left XRD indicates that the MAPb(Br0.6I0.4)3 film has not been soaked by light,the red line represents the sample soaked for 5 minutes in white light at ~50 mW·cm–2,and the blue line indicates that it is placed in the dark state 2 hour.On the right,by turning on(457 nm,15 mW·cm–2)and turning off the excitation light(after 5 minutes in the dark),the spectrum can be repeated between two states[86].(b)Reversible conversion between MAPbI3 and PbI2 under electric field.On the left is a cross-sectional view of lateral MAPbI3 perovskite solar cell in which PbI2 line is formed below the anode.The image to the right shows a schematic representation of the lateral MAPbI3 device with the PbI2 line moving toward the cathode,which is caused by two sets of solid-state chemical reactions that occur at the “A/B” and “Region B/C” interfaces,respectively[35].(c)SEM top view of the perovskite transverse structure with silver electrodes before(left)and after polarization(right).The “+” and “–” symbols indicate different electrode polarizations[71].
其次,對晶界和鈣鈦礦薄表面缺陷進行鈍化可以有效抑制離子遷移[122,123].如目前應用最廣泛的有機電子傳輸材料富勒烯及其衍生物PCBM,就可對鈣鈦礦晶界和表面缺陷進行鈍化進而有效地抑制離子遷移.此外,Wei等[29]的研究進一步指出,結合 polyethylene glycol(PEG)對薄膜形貌的改善和PCBM對缺陷的鈍化作用,可有效抑制離子遷移,提高器件性能.多項研究已表明,具有“倒置”平面結構并覆蓋有富勒烯電子傳輸層的器件與具有介孔結構的鹵化物鈣鈦礦太陽能電池比較,通常表現出更小的電流遲滯現象.Bi等[124]的研究指出該現象是因為沉積在鈣鈦礦薄膜上的富勒烯會擴散進入鈣鈦礦的晶界,并對晶界和粒子表面中的電荷陷阱進行鈍化,從而有效的抑制了電流遲滯現象.Xu等[73]通過進一步研究表明,鈣鈦礦薄膜晶界處存在深能級陷阱I-Pb反位(由I原子占據的Pb位置)缺陷,而PCBM與I-Pb反位缺陷之間有很強的電荷轉移能力,可以將深能級陷阱轉化為導帶以下的淺能級陷阱,如圖11(b).Zhao團隊[75]通過對比在鈣鈦礦膜表面覆蓋PEG或PCBM三種材料,發現PCBM與鈣鈦礦薄膜表面I–離子具有較強的相互作用從而可有效地鈍化表面I缺陷,抑制鈣鈦礦薄膜表面離子遷移,如圖11(c).此外,Zhao 團隊[125]研究了將醋酸銫(Cs Acetate)作為阻擋層旋涂在鈣鈦礦與Sprtio-OMeTAD之間,形成三明治結構,有效抑制了MA+向有機傳輸層的遷移,消除對有機傳輸層的破壞,提高了器件的運行穩定性.在他們的研究中指出,離子遷移對有機傳輸層具有較強的破壞作用,會嚴重影響器件的長期穩定性[30].
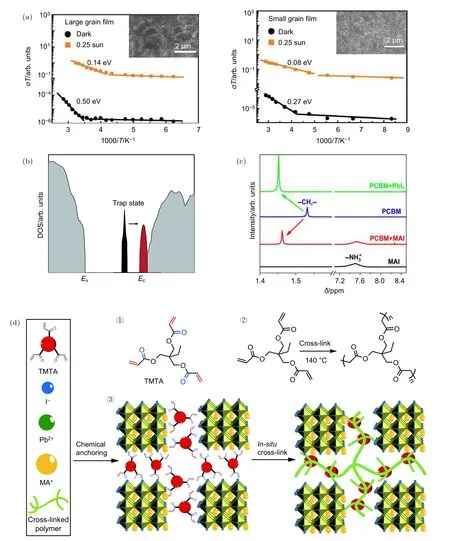
圖11 (a)大晶粒尺寸(左)和小晶粒尺寸(右)薄膜的溫度依賴離子電導性圖[119];(b)狀態密度(DOS)的 DFT 計算表明,Pb-I抗靜電缺陷引起的深陷阱態(黑色)減少,并且當PCBM吸附在有缺陷的鹵化物上時變得更淺(紅色)[73];(c)NMR圖示,PCBM 中特征峰—CH2—在 PCBM+MAI和 PCBM+PbI2中均有位移,表明碘和 PCBM 之間的相互作用[75];(d)原位交聯有機/鈣鈦礦薄膜的示意圖,①中具有顯著羰基(藍色)和烯基(紅色)基團的TMTA的化學結構,②在熱條件下TMTA的交聯聚合,③TMTA化學錨定到MAPbI3的晶界,然后原位交聯到連續的網絡聚合物[126]Fig.11.(a)Temperature-dependent ion conductivity of large grain size(left)and small grain size(right)films figure[119].(b)DFT calculations show that the deep trap state(black)caused by Pb-I antistatic defects is reduced and becomes lighter(red)when PCBM is adsorbed on the defective halide[73].(c)NMR shows that the characteristic peak —CH2— in PCBM is shifted in both PCBM+MAI and PCBM+PbI2,indicating the interaction between iodine and PCBM[75].(d)Schematic diagram of in situ crosslinking of organic/perovskite films:① Chemical structure of TMTA having significant carbonyl(blue)and alkenyl(red)groups;② crosslinking polymerization of TMTA under hot conditions;③ TMTA is chemically anchored to the grain boundaries of MAPbI3 and then crosslinked in situ to the continuous network polymer[126].
引入有機分子鈍化晶界也是抑制離子移動的有效手段.Li等[126]在鈣鈦礦薄膜中引入可交聯的液體有機小分子 trimethylolpropane triacrylate(TMTA),借助TMTA與PbI2的配位作用釘扎在晶界處,如圖11(d),同時 TMTA 經過熱處理,在晶界處發生原位交聯形成穩定的交聯的聚合物網絡,鈍化了晶界缺陷,離子遷移活化能EA也由0.21 eV 提升至 0.48 eV.此外,通過引入大分子,利用其與有機陽離子(MA+)之間較強的 π 鍵作用力,可有效抑制有機陽離子的遷移.如Wei等[127]通過DFT計算,發現鈣鈦礦中紅熒烯(rubrene)與有機陽離子之間的超分子陽離子- π 鍵相互作用能量(~1.5 eV)遠大于有機基陽離子遷移活化能(如 MA+,0.5—0.8 eV),從而有效抑制了 MA+離子遷移.該研究采用TOF-SIMS測試驗證了即使在偏壓(1 V 直流)、高溫(90 ℃)、持續光照(72 h)的環境下,有機陽離子與紅熒烯形成的超分子陽離子- π 鍵作用仍有效地抑制有機陽離子的遷移.
5.2 組分調控
鈣鈦礦材料是一種松散的離子晶體,原則上增加A,B,X位離子價態可以從本質上提高鈣鈦礦穩定性[90].增加離子價態主要是增加了離子與晶體框架間的庫侖相互作用,進而增加離子遷移活化能.Pan等[128]研究了具有雙鈣鈦礦結構的Cs2AgBiBr6單晶,其中 Pb2+離子被 Ag+和 Bi3+離子取代.相比之下,Cs2AgBiBr6顯示出 0.348 eV的離子遷移活化能,幾乎是 MAPbBr3(0.126 eV)的 3倍,DFT理論計算進一步表明,擴散勢壘VBr在 Cs2AgBiBr6(0.33 eV)中比在 MAPbBr3(~0.2 eV[129])大,證實 Cs2AgBiBr6中離子遷移受到抑制.然而,Cs2AgBiBr6(2.1 eV)的寬帶隙限制了對太陽光的有效利用,導致轉換效率低下.目前,關于高價位的陽離子鈣鈦礦材料的研究仍處于初步階段,與Pb2+陽離子相比,其電子結構和相穩定性較差[130],因而限制了其進一步的發展.
Mosconi和 de Angelis[131]研究認為,極性有機陽離子MA+旋轉引起的屏蔽效應可能會削弱移動離子與帶電鈣鈦礦晶格間的庫侖相互作用,進而加速離子遷移.而且有研究表明MA+離子的遷移會引起鈣鈦礦的結構可逆膨脹-收縮,最大晶格膨脹或收縮可達約4.4%[44].因此,選用極性較小、可旋轉性較差的A位陽離子(FA+和Cs+)替代MA+可以減緩鈣鈦礦中的離子遷移[132,133].DFT理論計算表明FAPbI3會抑制離子的移動,I–離子遷移活化能由 MAPbI3中的 0.32 eV增加到FAPbI3中的 0.55 eV[134].Zhang 等[94]通過實驗分別對MAPbI3器件和FAPbI3器件進行穩定性測試,如圖12(a),該研究結果表明在約20%濕度的暗態環境中,MAPbI3器件 10 d后完全降解而FAPbI3器件在大約一個月后才完全降解.此外,Zhou等[135]在 CsPbI2Br和 MAPbI3薄膜中定量研究了光照對離子遷移的影響.與MAPbI3薄膜隨著光強增大離子遷移活化能減小不同,CsPbI2Br薄膜在不同光強下離子遷移活化能不變,為固定值(~0.45 eV),如圖12(b),這將有利于開發持續光照下高穩定性的鈣鈦礦太陽能電池.
進一步研究發現,調整A位陽離子MA+,FA+,Cs+共混比例,可以有效提高鈣鈦礦結構的穩定性和抑制離子遷移.Li等[136]根據容忍因子,調整FA+,Cs+共混比例,獲得了穩定的鈣鈦礦結構.該研究表明,與純 FAPbI3(~165 ℃)和 CsPbI3(>300 ℃)相比,FA1–xCsxPbI3共混體系(<125 ℃)顯示出較低的d-a相轉變溫度,如圖12(c),而且在高濕度環境中不會發生a-d的相變.同樣基于純FAPbI3易轉變成d相,也可以通過用MA+部分置換FA+在室溫下達到穩定的a相[137].此外,Cs+部分取代FA+可以避免相分離并產生具有更寬帶隙(~1.74 eV)的混合鈣鈦礦,可用于疊層鈣鈦礦太陽能電池[138].
盡管根據容忍因子公式計算分析,堿金屬離子如 Li+,K+,Na+等離子因半徑小,容忍因子不在形成穩定鈣鈦礦相的數值區間范圍內(0.8—1.0[136]),即其不能單獨與PbX6八面體形成穩定鈣鈦礦相.但 K+離子[139]和 Na+離子[140]少量摻雜在 CIGS中的成功應用還是引起了人們在鈣鈦礦薄膜中摻雜堿金屬離子的興趣.研究表明,在鈣鈦礦中少量摻雜堿金屬離子,也可以消除遲滯現象,提高器件穩定性[141?143].Wang 等[144]提出 KCl預處理工藝,觀察到在熱退火過程中K+和Cl–離子會擴散到鈣鈦礦膜中,并在到達體缺陷時停止擴散進行鈍化作用.另有研究表明,KCl保留在界面功能層上且有效地鈍化缺陷,顯著地抑制SnO2/鈣鈦礦界面處的復合[143].在另一項研究中,Nam 等[142]摻入 K+部分取代Cs+,平均PCE由8.2%增加到9.1%,而且空氣環境下(20 ℃,20% RH)經過 120 h 仍保持初始效率的 80%,相比之下 CsPbI2Br經過 70 h只能達到其初始效率50%.Li等[145]通過進一步研究,將堿金屬離子(Li+,Na+,K+和 Rb+)摻雜到CsPbBr3鹵化物中,結構表征揭示由于堿金屬離子的加入,鈣鈦礦立方體積收縮,導致晶粒尺寸增大,如圖12(d)并且抑制鈣鈦礦膜中的載流子復合.
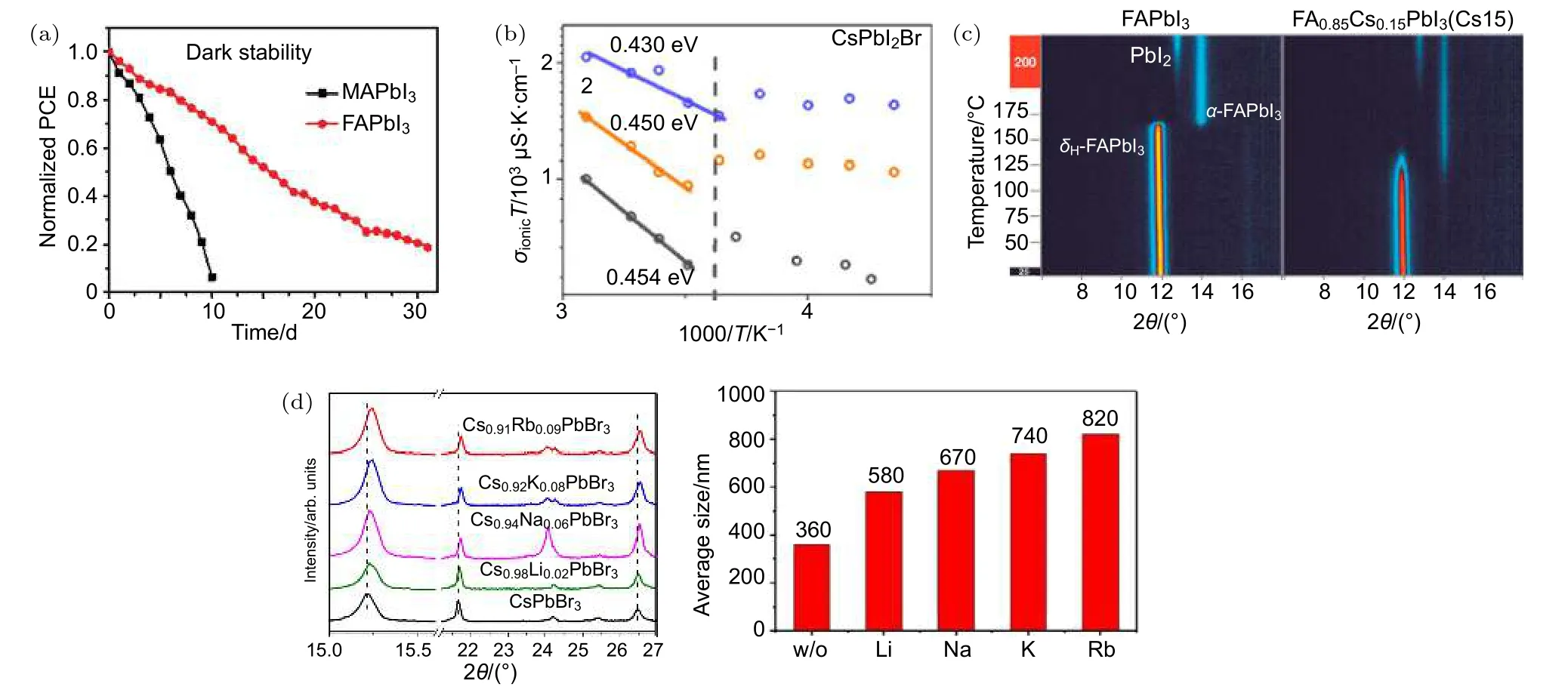
圖12 (a)MAPbI3(黑色方塊)和FAPbI3 (紅色圓圈)裝置下暗穩定性(~20%相對濕度(RH))[94];(b)在不同光強下,CsPbI2Br薄膜的溫度依賴離子電導性圖,擬合得離子遷移活化能為常數(~0.45 eV)[135];(c)FA1–xCsxPbI3的溫度依賴性XRD圖,純 FAPbI3的 d-a 相轉變在約 165 ℃ 發生,摻 15% Cs則 d-a 相轉變溫度降低至約 125 ℃[136];(d)CsxR1–xPbBr3(R=Li+,Na+,K+和Rb+)的XRD圖(左)及平均晶粒大小圖(右)[145]Fig.12.(a)Dark stability(~20% RH)of MAPbI3(black squares)and FAPbI3(red circles)devices[94];(b)the ion mobility activation energy of CsPbI2Br film is constant(~0.45 eV)under different light intensities[135];(c)the temperature-dependent XRD pattern of FA1–xCsxPbI3,the d-a phase transition of FAPbI3 occurs at about 165 ℃,and the d-a phase transition temperature is reduced to about 125 ℃ when 15% Cs is doped[136];(d)XRD pattern(left)and average grain size map(right)of CsxR1–xPbBr3(R=Li+,Na+,K+ and Rb+)[145].
5.3 晶體維度調控
二維層狀鈣鈦礦早在1980年代末就已被提出,但直到近幾年研究表明二維層狀鈣鈦礦相比于三維鈣鈦礦具有更好的穩定性才引起研究者們的普遍關注[138,146?152].Lin等[153]首先報道了二維結構對于抑制離子遷移的優勢,該研究通過對三維MAPbI3和n=4的二維層狀鈣鈦礦(BA)2(MA)3Pb4I13進行與溫度依賴性電導測試研究,發現二維層狀鈣鈦礦在330 K以下幾乎沒有離子遷移現象.相比之下,在三維鈣鈦礦中超過260—290 K時,離子電導就開始主導薄膜的導電性,該現象與Yuan等[43]的研究一致.Xiao等[154]的研究進一步證實,層狀鈣鈦礦BA2MA2Pb3I10(n=3)中沿面內方向的離子遷移會被抑制.該研究表明及在準2D鈣鈦礦中的形成能均比3D鈣鈦礦中形成能高,導致準2D鈣鈦礦中空位密度相對于3D鈣鈦礦低得多,因此準2D鈣鈦礦本質上在電場下具有更好的穩定性.事實上,低維結構的鈣鈦礦在發光器件中發現了更多應用,其對防止離子遷移具有更高的要求[155?159].
然而,目前低維鈣鈦礦雖然具有較好的穩定性優勢,但是在性能上仍然低于3D鈣鈦礦,仍有較大的發展空間[160].Zhang等[161]通過在準2D鈣鈦礦(BA)2(MA)3Pb4I13中摻雜少量Cs元素,電池效率提高到了13.7%.同時,準2D鈣鈦礦穩定性也顯著提升,30%濕度情況下,該器件經過1400 h后,效率還保持初始效率的89%.同時Cs元素的加入有利于控制晶向,促進晶粒尺寸增長.此外,在3D鈣鈦礦上旋涂2D鈣鈦礦材料,形成2D-3D結構的鈣鈦礦薄膜具有更好的穩定性和更高的器件性能[162?166].如經 AVAI/IPA(5-aminovaleric acid iodide)溶液處理,經熒光光譜分析,沉積AVAI溶液形成的2D鈣鈦礦覆蓋在3D鈣鈦礦之上,鈍化了3D鈣鈦礦表面缺陷.相應的2D/3D結構器件PCE達到18%,并且在空氣環境中20 d可保持初始效率的72%[150].此外,研究者還通過使用共軛有機基團作為陽離子[167,168],進一步提高準2D鈣鈦礦太陽能電池的效率與電荷傳輸能力.
6 總結與展望
雖然金屬鹵化物鈣鈦礦太陽能電池正在快速發展,但是實現商業化仍面臨著若干關鍵挑戰.制備大面積、高效率和長期穩定的鈣鈦礦器件,以及尋找更好的能取代有毒鉛離子,發展非鉛鹵化物鈣鈦礦太陽能電池仍然是未來發展的主要方向.本綜述詳細闡述了鹵化物鈣鈦礦材料中的離子遷移現象.對鹵化物鈣鈦礦材料中發生離子遷移的離子種類及這些離子的形成條件做了深入詳細的討論.深入理解離子遷移現象及其對器件性能和長期穩定性的影響機制,對于鹵化物鈣鈦礦太陽能電池的發展和應用至關重要.尋找有效的抑制甚至消除離子遷移的方法,對于器件的長期穩定性具有重要作用.此外,離子遷移并非只對器件帶來不利影響.在特定條件下,利用離子對鈣鈦礦薄膜的化學摻雜效應、自建電場的調節功能,設計具有獨特功能或光電響應行為的器件也是值得關注的研究方向.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
