殼聚糖-海藻酸鈉載藥微球制備工藝研究
2019-09-06 03:22:34史同瑞崔宇超王麗坤朱慶賀楊旭東張艷高俊峰
中國獸藥雜志 2019年8期
關鍵詞:殼聚糖
史同瑞,崔宇超,王麗坤,朱慶賀,楊旭東,張艷,高俊峰
(黑龍江省農業科學院畜牧獸醫分院,黑龍江齊齊哈爾 161005)
微球(microspheres,MS)是指將藥物溶解、分散、吸附或包裹在高分子聚合物的載體材料中,并制成載藥的球狀微粒。微球粒徑一般是在1~1000 μm范圍內,常見的微球粒徑為1~40 μm,小于1 μm的微球稱為納米微球[1]。微球作為一種載物體系,已在藥物載體、酶固定化、細胞培養微反應器、基因運載等方面開展了較為深入的研究[2,3]。微球作為藥物的包封、緩釋、控釋與靶向釋放的藥物載體,具有保護敏感藥物成分免受胃腸道環境破壞,提高藥物穩定性,延長藥物作用時間,提高藥物的生物利用度等許多優點,因此,微球作為藥物的載體具有廣闊的開發應用前景[4-6]。
微球多采用明膠、殼聚糖、海藻酸鈉等可生物降解的高分子無毒天然材料制備而成,制備方法常采用滴注法、噴霧法、原位聚合法、靜電法和乳化法等方法,然而這些方法均存在著一定的弊端:一是需要使用化學有機溶劑,這不僅會造成毒性物質殘留等問題,而且還可能導致藥物喪失活性;二是需要復雜的生產設備,生產效率低,這給載藥微球產業化生產造成了障礙[7],研究開發安全、簡便、高效的生產方法是微球制劑實現產業化的技術需求。本試驗結合微乳及膠體制備工藝[8],采用D相乳化法,以硫酸小襞堿為模型藥物,以藥物包封率為評價指標,應用響應面法篩選優化了微球的制備工藝,并制備了優良的硫酸小襞堿殼聚糖-海藻酸鈉微球,這為微球制劑的產業化開發奠定了一定的技術基礎。
1 材料與方法
1.1 材料 殼聚糖,分析純,脫乙酰度DD%>85%,批號170902,武漢合中生化制造有限公司;海藻酸鈉,分析純,批號S817372,上海麥克林生化科技有限公司。硫酸小襞堿,含量98.21%,批號180301,四川恒瑞通達生物科技有限公司;硫酸小檗堿標準品,含量98.7%,中國獸醫藥品監察所;氯化鈣,分析純,天津市恒興化學試劑制造有限公司;液體石蠟,批號20160905,天津市巴斯夫化工有限公司;丙二醇、正丁醇和正戊醇,液體石蠟、司班-80、吐溫-80,均為分析純,天津市巴斯夫化工有限公司。
1.2 儀器 UV1900PC型雙光束紫外可見光分光光度計,上海佑科儀器儀表有限公司;winner802納米粒度儀,濟南微納顆粒儀器股份有限公司;ZNCL-S型恒溫磁力攪拌器,上海羌強儀器設備有限公司;MS104TS/02型電子分析天平,梅特勒-托利多有限公司。
1.3 微球制備方法 精密稱取海藻酸鈉2 g,硫酸小襞堿4 g,加入100 mL蒸餾水中,充分溶解,混合均勻;取制備的海藻酸鈉-硫酸小襞堿混合液100 mL,加入液體石蠟50 mL,司班-80 5 mL,吐溫-80 10 mL,丙二醇5 mL,以800 r/min充分攪拌乳化,制成含有海藻酸鈉及藥物的水包油型乳膠。取適量乳膠,加入等量1%氯化鈣溶液中,以8000~1000 r/min高速攪拌,膠凝反應一定時間,過濾得微球;按著微球與殼聚糖溶液體積比1∶10比例將微球加入2%殼聚糖溶液中,再次膠凝1 h,蒸餾水洗滌,過濾,干燥,即得載藥微球。
1.4 微球評定指標測定
1.4.1 硫酸小檗堿含量測定
1.4.1.1 硫酸小檗堿標準品溶液制備 精密稱取硫酸小檗堿標準品0.1 g,置于100 mL容量瓶中,加入20 mL去離子水,充分震蕩至完全溶解,再加去離子水至刻度,搖勻,制成硫酸小檗堿濃度為1000 μg/mL的標準品儲備液。精確量取標準品儲備液0.05、0.1、0.2、0.5、1.0、2.0、5.0 mL分別置于100 mL容量瓶中,加水定容至刻度,搖勻,制成硫酸小檗堿濃度分別為0.5、1.0、2.0、5.0、10.0、20.0、50.0 μg/mL的標準溶液。
1.4.1.2 標準曲線繪制 取各濃度硫酸小檗堿標準液樣品,以去離子水作空白對照。參照文獻方法[9]在423 nm波長處測定其吸光度,各濃度標準液樣品重復測定3次。以硫酸小檗堿濃度為橫坐標(x),吸光度值為縱坐標(y)進行線性回歸,繪制標準曲線,計算回歸方程。
1.4.2 微球評定指標測定
1.4.2.1 藥物包封率測定 以藥物包封率作為微球制備工藝的評定指標。取制備的微球懸液用0.22 μm濾膜過濾,并用適量去離子水沖洗3次。取濾液及沖洗液混合液,按照1.4.1方法于423 nm波長處測定藥物吸光度,計算混合液中硫酸小檗堿含量及微球藥物包封率[10]。包封率(EE%)=微球中藥物量/總投藥量×100%。
1.4.2.2 微球形態與粒徑測定 取硫酸小襞堿殼聚糖-海藻酸鈉微球樣品置于載玻片上,用顯微鏡觀察微球形態。應用納米粒度儀測定微球的大小及其粒徑分布。
1.5 制備工藝單因素試驗 在單因素預選試驗基礎上,確定海藻酸鈉溶液濃度、氯化鈣溶液濃度、殼聚糖溶液濃度,以及初次膠凝時間是影響微球質量的主要因素。根據預試驗結果,每個因素選擇5個水平進行單因素試驗。
1.5.1 海藻酸鈉溶液濃度篩選 在氯化鈣溶液濃度2.0%、殼聚糖溶液濃度1.2%、初次膠凝時間30 min條件下,以海藻酸鈉溶液濃度分別為0.5%、1%、1.5%、2%、2.5%水平制備載藥微球,觀察微球形態,測定微球藥物包封率,確定海藻酸鈉溶液最適濃度。試驗重復3次。
1.5.2 氯化鈣溶液濃度篩選 在海藻酸鈉溶液濃度1.5%、殼聚糖溶液濃度1.2%、初次凝膠時間30 min條件下,以氯化鈣溶液濃度分別為1.0%、1.5%、2.0%、2.5%、3.0%水平制備載藥微球,觀察微球形態,測定藥物包封率,確定氯化鈣溶液最適濃度。試驗重復3次。
1.5.3 初次凝膠時間篩選 在海藻酸鈉溶液濃度1.5%、氯化鈣溶液濃度2.0%、殼聚糖溶液濃度1.2%條件下,以初次凝膠時間分別為10、20、30、40、50 min水平制備載藥微球,觀察微球形態,測定藥物包封率,確定初次凝膠最適時間。試驗重復3次。
1.5.4 殼聚糖溶液濃度 在海藻酸鈉溶液濃度1.5%、氯化鈣溶液濃度2.0%、初次凝膠時間30 min條件下,分別以為殼聚糖溶液濃度0.4%、0.8%、1.2%、1.6%、2%水平制備載藥微球,觀察微球形態,測定微球藥物包封率,確定殼聚糖溶液最適濃度。試驗重復3次。
1.6 制備工藝優化
1.6.1 試驗設計 基于單因素試驗結果,應用Box-Behnken設計原理,設計四因素三水平響應曲面分析試驗,優化載藥微球制備的各因素最適水平,因素水平設計見表1。
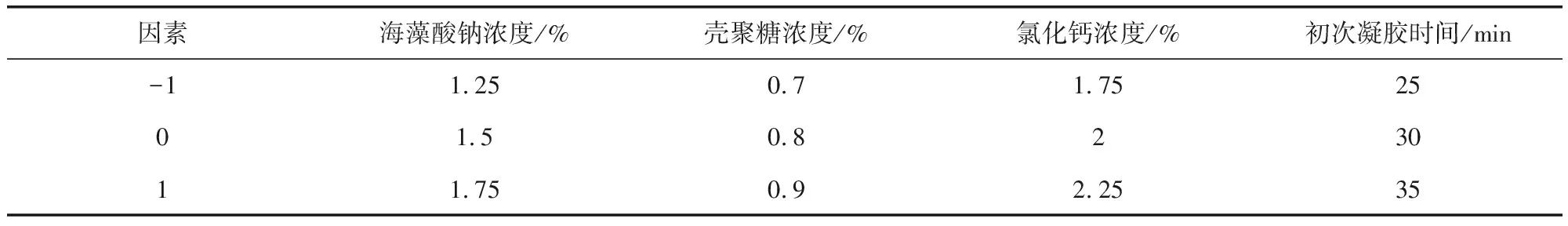
表1 Box-Behnken設計的因素水平Tab 1 The factors and levels for Box-Behnken design
1.6.2 數據處理及模型擬合 應用Design Expert 8.0.6軟件中Box-Behnken方法,對制備硫酸小檗堿微球所得數據進行處理,將藥物包封率(因變量)與各因素(自變量)進行多元線性回歸和二項式方程擬合,響應曲面法設計方案見表2。
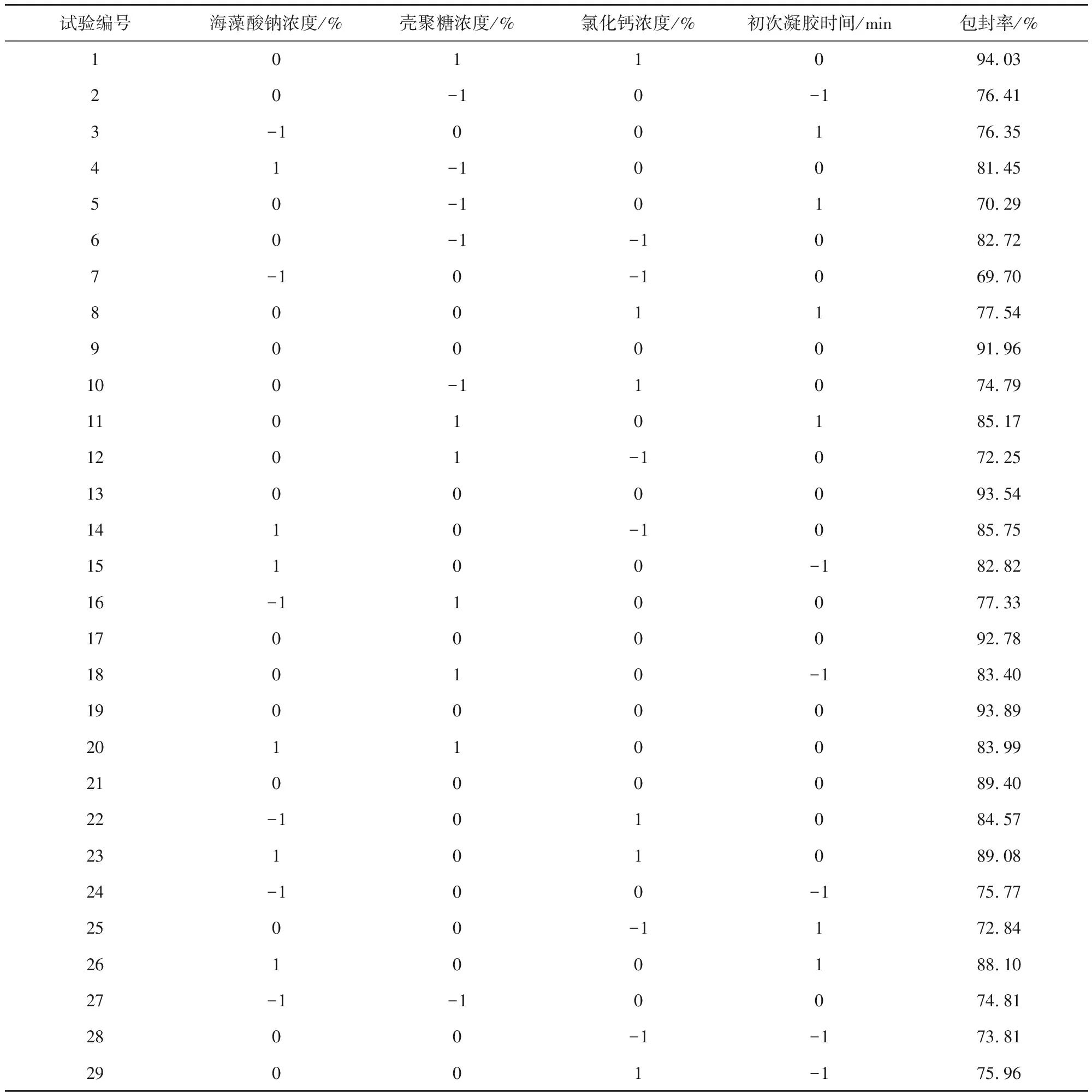
表2 響應曲面法設計方案Tab 2 Response surface methodology design scheme
1.6.3 驗證試驗 根據優化的海藻酸鈉溶液濃度、殼聚糖溶液濃度、氯化鈣溶液濃度,以及初次凝膠時間,按照制備工藝流程,測定微球的藥物包封率,并進行3次驗證試驗,驗證預測結果的準確性。
2 結果與分析
2.1 標準曲線繪制 以吸光度值為縱坐標,硫酸小檗堿濃度為橫坐標繪制標準曲線,得出回歸方程:y=0.0129x+0.0394(R2=0.999)。由圖1可知,硫酸小檗堿質量濃度在0.5~50 μg/mL范圍內時,其吸收度與藥物濃度的線性關系良好。
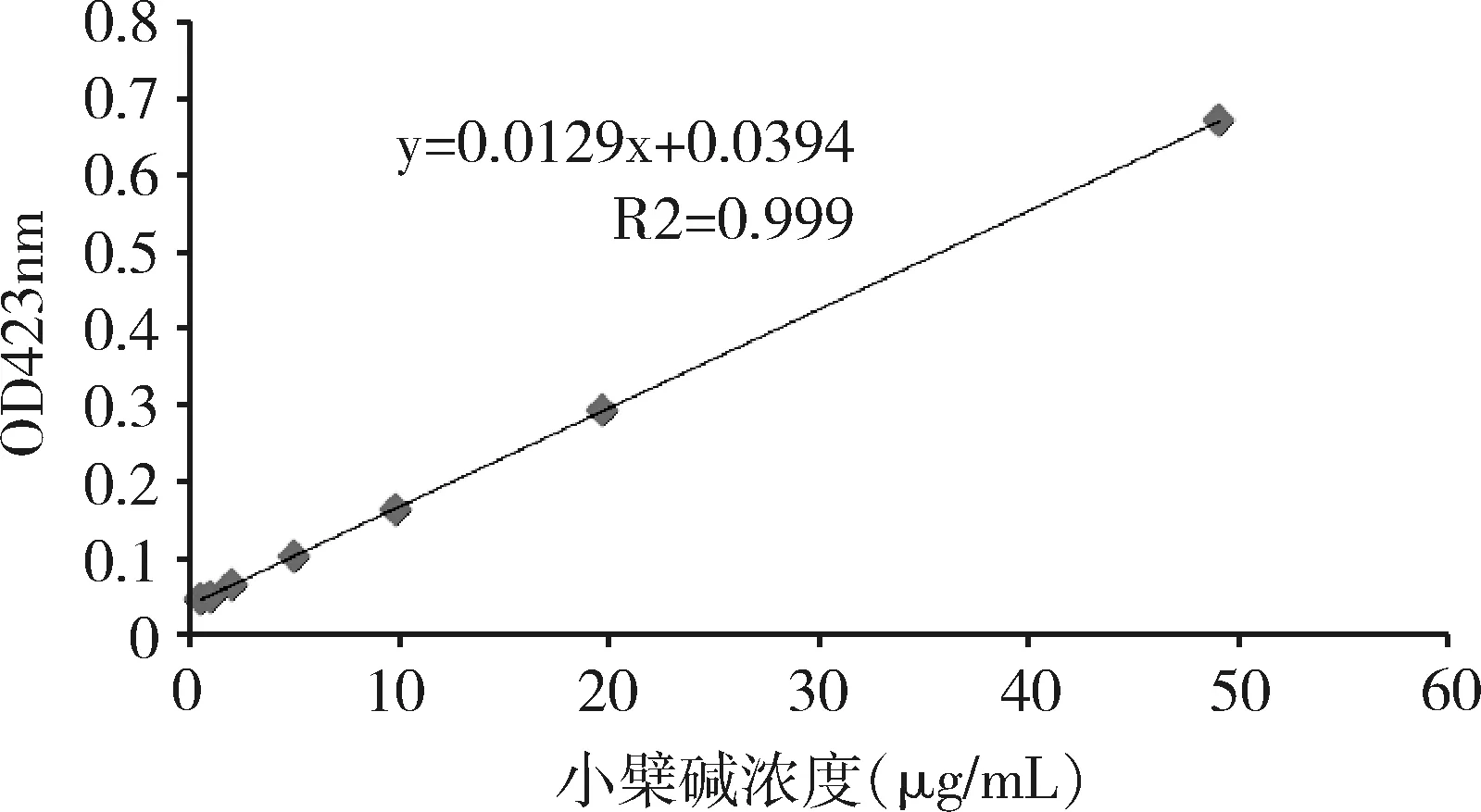
圖1 硫酸小檗堿標準曲線Fig 1 Standard curve of Berberine Sulfate
2.2 單因素試驗結果
2.2.1 海藻酸鈉溶液濃度對微球性能的影響 由圖2可知,隨著海藻酸鈉溶液濃度的增大,微球藥物包封率逐漸升高,整齊度逐漸變好,粒徑無明顯變化。當海藻酸鈉濃度超過2.0%時,雖然微球的包封率也逐漸升高,但微球的圓整度下降,制備微球難度加大。綜合考慮各因素,選擇海藻酸鈉溶液的最佳濃度為1.5%。
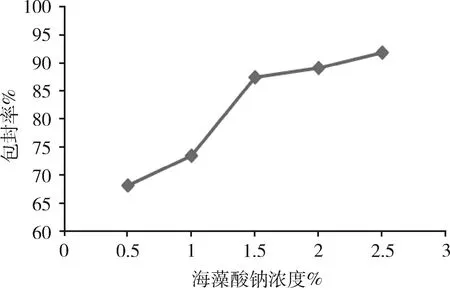
圖2 海藻酸鈉濃度對微球包封率的影響(n=3)Fig 2 Effect of the sodium alginate concentration onencapsulation efficiency of microspheres
2.2.2 氯化鈣溶液濃度對微球性能的影響 由圖3可知,隨著氯化鈣溶液濃度的升高,藥物的包封率也逐漸升高,但當氯化鈣溶液濃度超過2%時,微囊的圓整度降低,脆性增大,因此,選擇氯化鈣溶液的最佳濃度為2%。
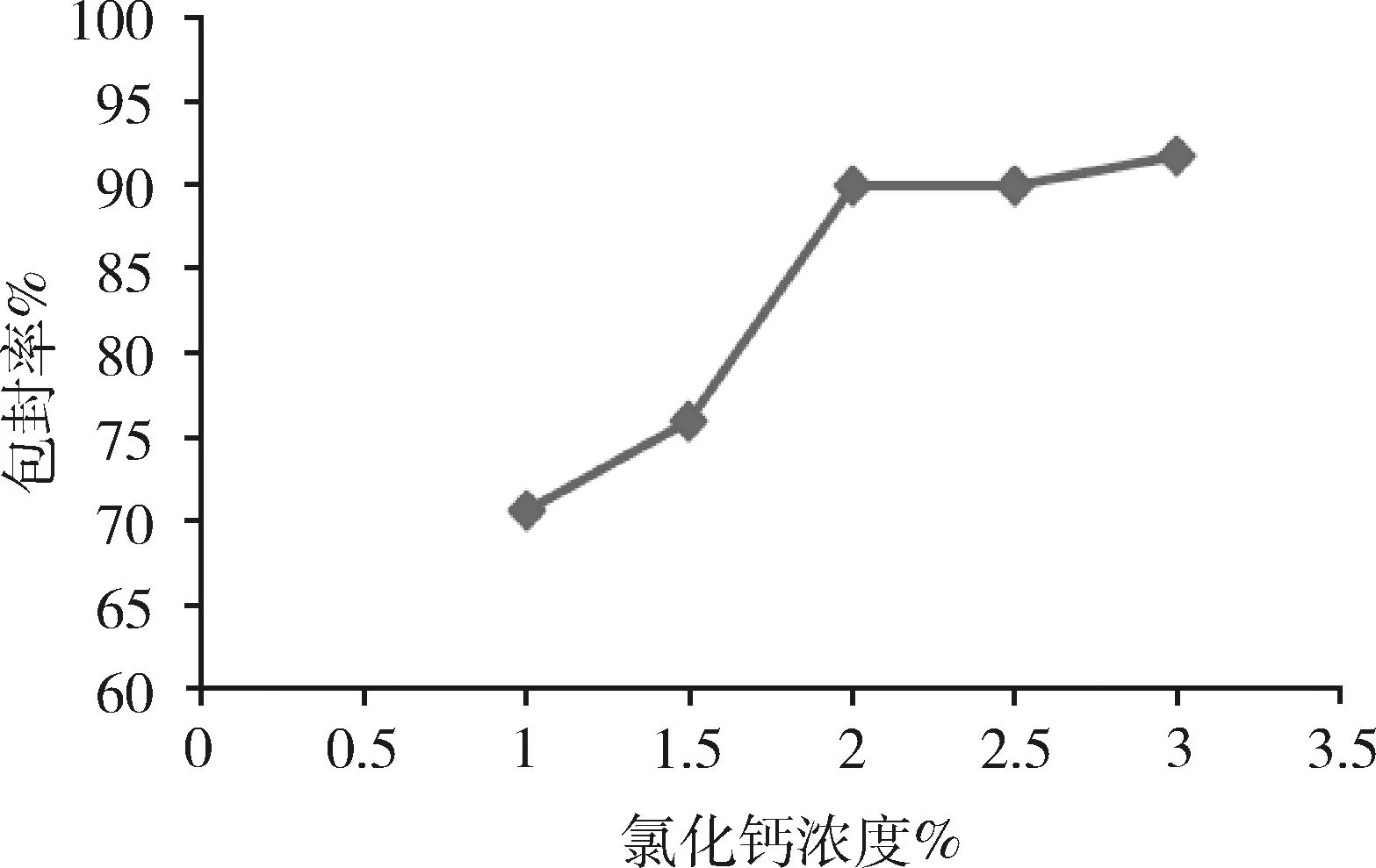
圖3 氯化鈣濃度對微球包封率的影響(n=3)Fig 3 Effect of the calcium chloride concentration onencapsulation efficiency of microspheres
2.2.3 初次凝膠時間對微球性能的影響 由圖4可知,隨著初次凝膠時間由10 min延長至30 min,微球的整齊度、藥物包封率逐漸升高,但初次凝膠時間由30 min延長至50 min時,微球的整齊度和藥物包封率變化不明顯(P>0.05),因此,選擇最佳初次凝膠時間為30 min。
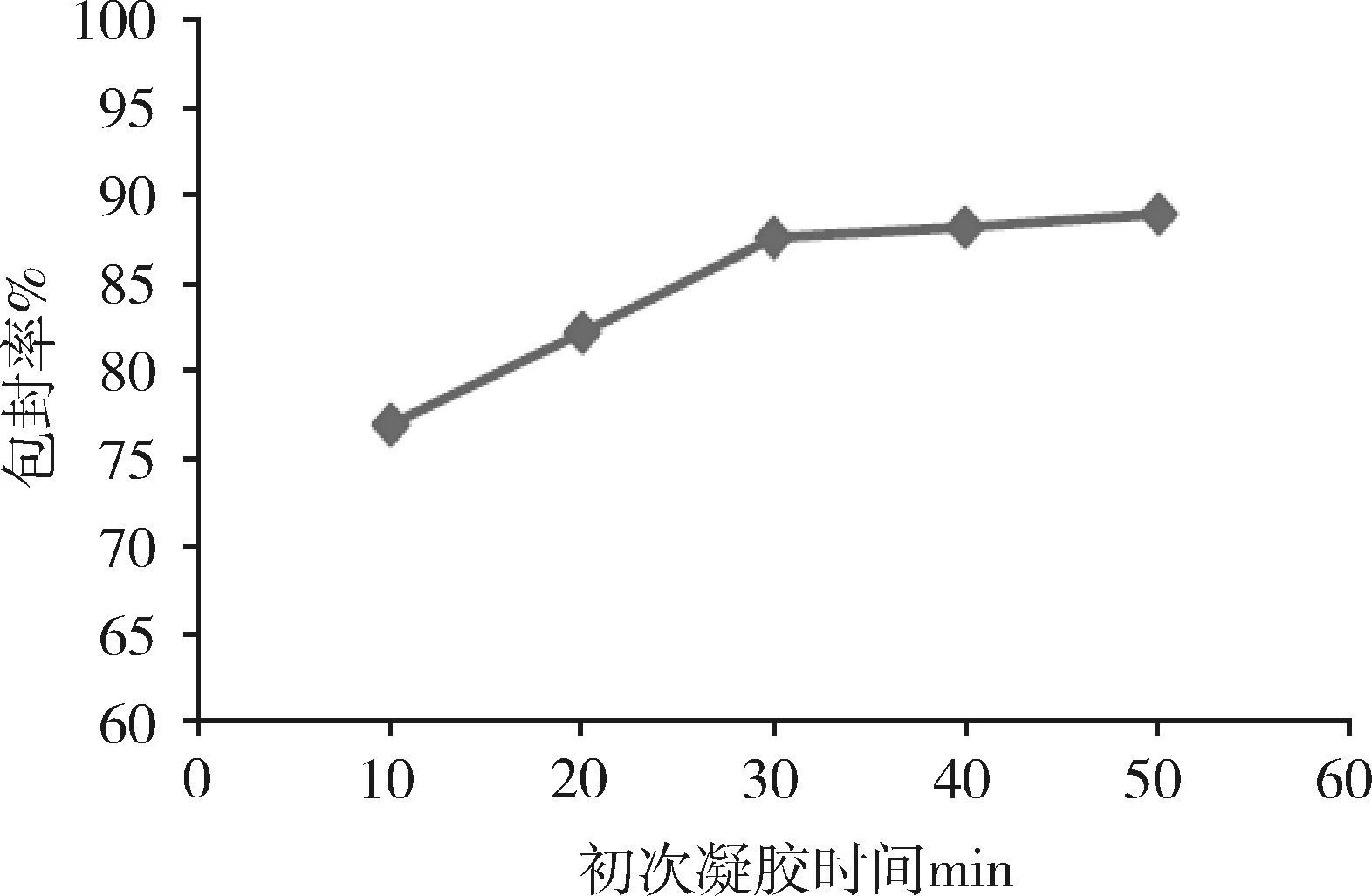
圖4 初次凝膠時間對微球包封率的影響(n=3)Fig 4 Effect of the gel-forming time onencapsulation efficiency of microspheres
2.2.4 殼聚糖溶液濃度對微球性能的影響 由圖5可知,隨著殼聚糖濃度增加,雖然微球的整齊度變化不明顯,粒徑也無明顯變化,但殼聚糖溶液濃度為0.8%時,微球的藥物包封率最高,因此,選擇殼聚糖溶液最佳濃度為0.8%。
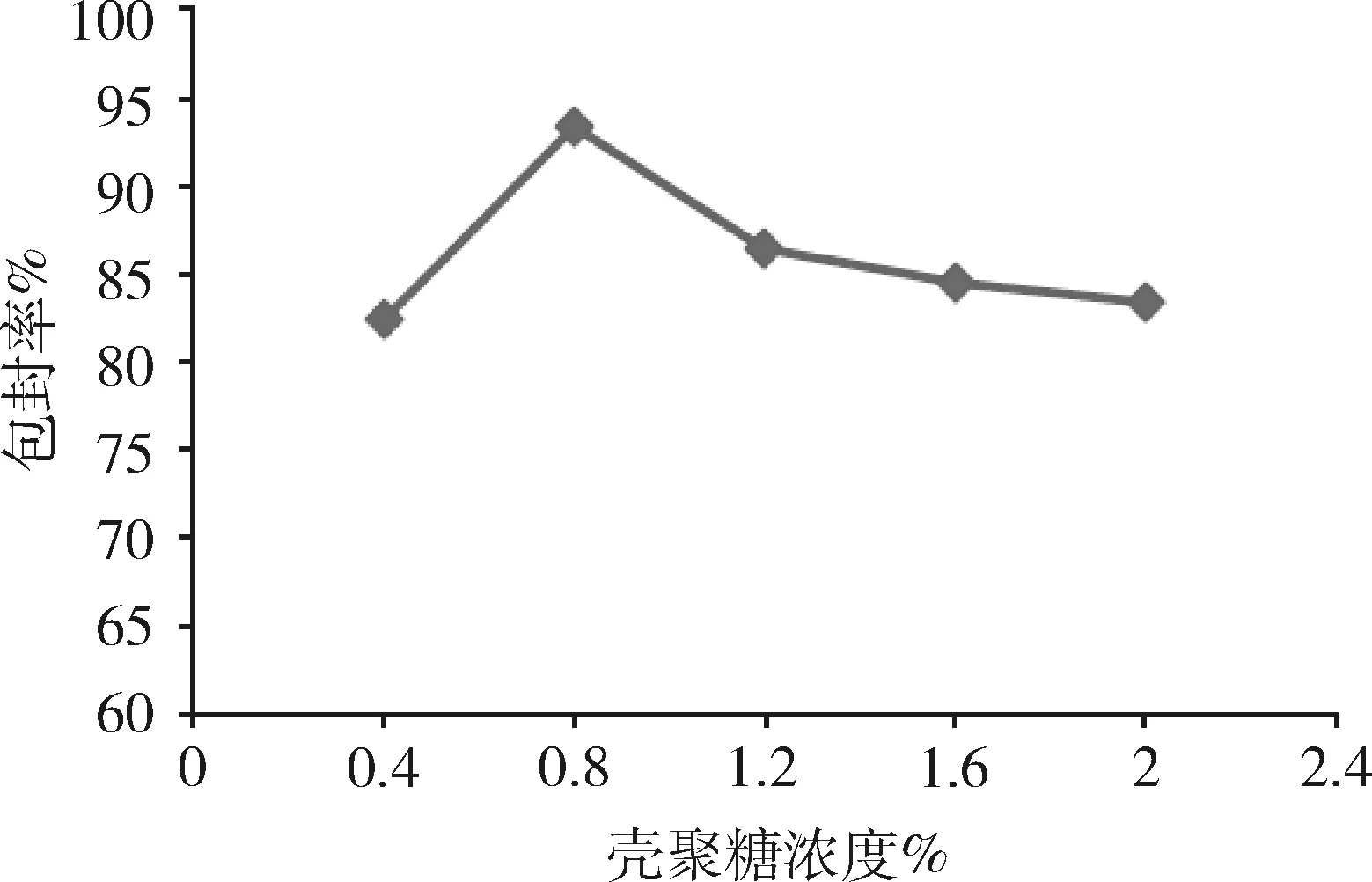
圖5 殼聚糖濃度對微球包封率的影響(n=3)Fig 5 Effect of the chitosan concentration onencapsulation efficiency of microspheres
2.3 制備工藝優化結果
2.3.1 統計模型與方差分析 通過回歸分析,得多元回歸方程:Y=92.31+4.39X1+2.97X2+3.24 X3+0.18X4+5.000E-003X1X2-2.88X1X3+1.18X1X4+7.43X2X3+1.97X2X4+0.64X3X4-4.48X12-6.12X22-6.57X32- 8.39X42,回歸系數R2=0.9059,回歸方程與實際模型擬合度良好。對二項式方程中各系數進行F檢驗,由表3可知,X1、X2、X3為極顯著因素,X2與X3的交互作用極顯著(P<0.01)。此模型的失擬項Pr>F值為0.0834,大于0.05,模型失擬項不顯著(P>0.05),說明模型選擇合理。
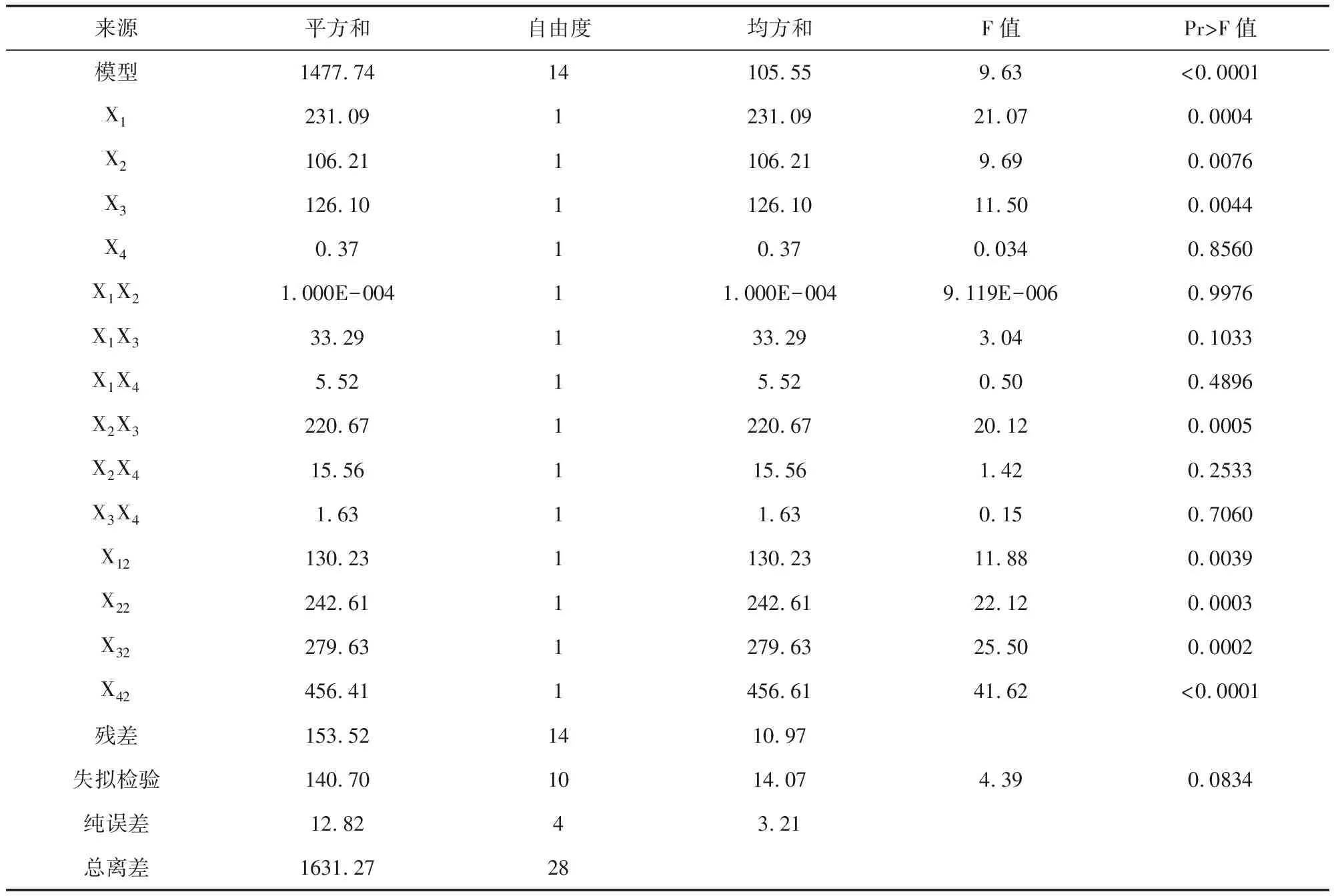
表3 回歸方程方差分析結果Tab 3 Variance analysis of regression equation
注:Pr>F值小于0.05,表明模型或考察因素有顯著影響;Pr>F值小于0.01,表明模型或考察因素有極顯著影響。
Notes: the value of Pr>F less than 0.05 indicates that the model or inspection factors have a significant influence; the value of Pr>F less than 0.01 indicates that the impact of the model or inspection factors is extremely significant.
2.3.2 響應曲面分析 利用Design Expert 8.0.6軟件作響應曲面圖及等高線圖,在篩選因素水平范圍內,微球藥物包封率存在最大值,即等高線圖標注的中心點。由圖6可知,當殼聚糖溶液濃度一定時,隨氯化鈣溶液濃度的升高,包封率快速增大,然后又減小。當氯化鈣溶液濃度一定時,隨著殼聚糖溶液濃度的升高,包封率快速增大,然后又減小。響應曲面圖的曲線比較陡峭,說明殼聚糖、氯化鈣兩因素的交互作用對包封率影響顯著(P<0.05),包封率的最大值應在氯化鈣濃度1.95%~2.15%和殼聚糖濃度0.80%~0.90%范圍內。由圖7可知,當初次凝膠時間一定時,隨著海藻酸鈉濃度的升高,包封率快速增大,然后又減小。當海藻酸鈉溶液濃度一定時,隨著初次凝膠時間的延長,包封率快速增大,然后又減小,包封率的最優值應在海藻酸鈉溶液濃度1.45%~1.65%和初次凝膠時間29~33 min范圍內。等高線圖呈橢圓形,說明殼聚糖、氯化鈣兩因素具有一定的交互作用。
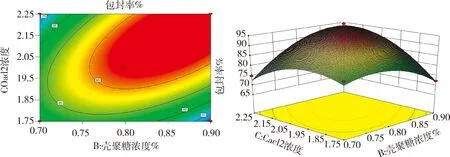
圖6 氯化鈣濃度與殼聚糖濃度交互響應面圖及其等高線圖Fig 6 Response surface and contour plots for the effect between calcium chloride concentration and chitosan concentration
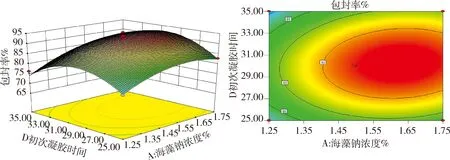
圖7 海藻酸鈉濃度與凝膠時間交互響應面圖及其等高線圖Fig 7 Response surface and contour plots for the effect between sodium alginate concentration and gel-forming time
2.3.3 優化分析 應用Design Expert 8.0.6軟件,模擬的最佳工藝條件:海藻酸鈉溶液濃度1.57%,殼聚糖溶液濃度0.86%,氯化鈣溶液濃度2.13%,初次凝膠時間為30.71 min,在此條件下,硫酸小檗堿微球包封率理論值為94.6991%。根據最優化工藝流程進行了3次驗證試驗,硫酸小檗堿包封率的實測平均值為94.09%,這說明試驗設計和數學模型具有可靠性和重現性。
2.4 微球形狀與粒徑分布 微球外觀形狀為規則球形,表面光滑圓整,無明顯的聚集現象,見圖8。納米粒度儀測定結果表明,應用篩選的最佳工藝制備的微球平均粒徑為329 nm,且呈正態分布,見圖9。
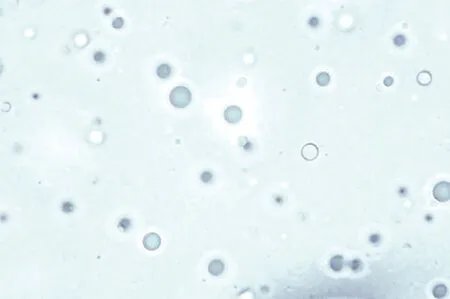
圖8 微球形態Fig 8 Morphology of microspheres
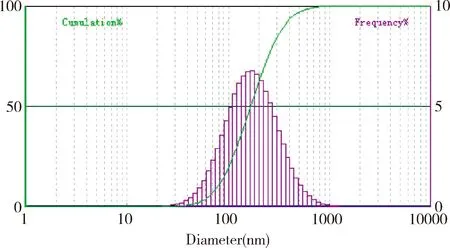
圖9 微球粒徑分布Fig 9 Distribution of particle sizes of microspheres
3 討論與結論
3.1 微球制備的影響因素 海藻酸鈉-殼聚糖載藥微球的形狀、致密度、藥物包封率、微球降解速率,以及藥物釋放動力學等質量性能,主要取決于制備微球的高分子材料及其濃度、交聯劑鈣離子濃度、聚合反應時間、溶液pH值等因素。由于采取的制備方法、生產的原材料和包載藥物的不同,因此微球制備的工藝條件也有差異。徐連敏等[11]研究表明,決定微球質量和藥物包封率的影響因素依次為殼聚糖濃度>氯化鈣濃度>海藻酸鈉濃度>藥物載量。張華等[12]確定的影響因素依次為海藻酸鈉質量分數>殼聚糖質量分數>氯化鈣質量分數>海藻酸鈉與藥物比。岳春華等[13]證實,影響微球質量的因素依次是明膠用量>藥料比>乳化劑用量>殼聚糖用量。本研究通過單因素試驗和響應曲面法證實,決定微球質量的主要影響因素依次為:海藻酸鈉溶液濃度>氯化鈣溶液濃度>殼聚糖溶液濃度>膠凝時間,從總體試驗結果看,本試驗與其他學者的研究結果基本一致,由此可見,這些因素是決定微球制備的關鍵因素。
3.2 工藝條件的篩選 在微球制備過程中,鈣離子作為交聯劑,首先與海藻酸鈉分子的-COO-結合形成凝膠球,凝膠球再與殼聚糖溶液混合,海藻酸鈉分子的羧基與殼聚糖分子的氨基絡合,形成致密的微球[14-15]。由于海藻酸鈉溶液、氯化鈣溶液及殼聚糖溶液濃度決定著溶液的粘度及其電荷數,所以溶液的濃度直接影響著微球的形成,在一定范圍內通過改變材料濃度可調節微球的質量特性。徐連敏等[11]研究表明,使用2%海藻酸鈉溶液、0.5%氯化鈣溶液和0.4%殼聚糖溶液,膠凝10 min,可制備理想微球。張華等[12]優化的制備工藝為2%海藻酸鈉溶液、2%氯化鈣溶液、0.1%殼聚糖溶液,海藻酸鈉與藥物比3∶1。岳春華等[13]篩選的最佳制備條件為2%明膠、1%殼聚糖、藥料比1∶1,所得微球成球性好、粒徑分布均勻。王家榮等[16]試驗證實,制備微球的適宜海藻酸鈉濃度為2%,氯化鈣濃度為1.5%。朱敏莉等[17]研究顯示,海藻酸鈉溶液濃度為3%時,能制成分散的大小整齊的圓形微球。張志辰等[18]研究認為,氯化鈣濃度為5%時,形成微球強度高、形狀規則、大小均勻。本研究表明,1.57%海藻酸鈉溶液、2.13%氯化鈣溶液、0.86%殼聚糖溶液和初次凝膠時間為30.71 min時,可自備藥物包封率最理想的微球。縱觀各研究結果,雖然不同學者制備微球的材料濃度均有一定的差異,筆者認為微球形成的機理主要是海藻酸鈉與鈣的交聯反應,以及海藻酸鈉與殼聚糖的聚合反應,由于材料分子的電荷及數量的確定性,其各材料間的質量分數比例理論上應基本一致,差異只是制備微球的材料溶液濃度而已。
3.3 包封率的影響因素 微球的藥物包封率是確定微球制備工藝優劣的重要指標之一。微球形成機理是在海藻酸鈉水溶液中加入鈣離子,海藻酸鈉分子鏈的古羅糖醛酸的鈉離子與鈣離子發生交換反應,形成相互交聯的網絡結構海藻酸鈣凝膠,海藻酸鈉分子再與殼聚糖分子絡合,最終形成具有機械強度和彈性的微球[19]。由此可見,海藻酸鈉濃度、殼聚糖濃度和氯化鈣濃度決定著微球的形成速度和致密度,從而影響著藥物的包封率。本研究表明,在一定濃度范圍內適度提高海藻酸鈉溶液和氯化鈣溶液的濃度,有利于提高微球的形成速度和藥物包封率,但過高的材濃度又會降低微球圓滑度等物理特性,從而影響影響微球的性能。在本研究篩選工藝條件下,制備微球的藥物平均包封率為94.09%,明顯高于何清義等[20]制備微球的包封率61%,與張華等[12]制備鞣花酸殼聚糖-海藻酸鈉微球的平均包封率97.73%,羅洋等[21]制備的硒太子參多糖納米微球包封率92.46%,以及徐連敏等[11]制備地塞米松磷酸鈉殼聚糖-海藻酸鈉微球包封率96.90%相近,這說明本試驗篩選工藝制備微球的質量良好。
3.4 微球制備方法的比較 依據微球制備工藝,微球的制備方法主要分為噴霧法和乳化法兩類。噴霧法需要特殊的生產設備,乳化法需要使用甲醛、丙酮等有機溶劑。這些方法不僅生產工藝復雜,生產效率低,而且使用的有機溶劑還存在著一定的毒性,污染環境,并可能導致藥物的失活,因此,現有的制備技術嚴重制約了微球藥劑的產業化進程[22]。本研究綜合D相乳化法,以及乳劑、微乳及膠體的制備方法[23],創新了微球的傳統制備方法和劑型。該方法首先制備不溶于水的乳膠,然后再利用乳膠在水中的不溶性,將乳膠依次加入氯化鈣和殼聚糖溶液中,乳化聚合制成載藥凝膠微球。本研究采用攪拌乳化法制備微球,克服了滴注法和噴霧法的弊端,不需要特殊的生產設備,生產工藝簡單,不使用有機溶劑,乳化及交聯條件溫和,制備微球整齊度好,藥物包封率高,這為微球制劑的產業化生產奠定了技術基礎。
3.5 結論 本試驗應用海藻酸鈉、殼聚糖可生物降解的無毒高分子材料,優化的硫酸小襞堿殼聚糖-海藻酸鈉微球制備工藝條件為:海藻酸鈉溶液濃度1.57%,氯化鈣溶液濃度2.13%,殼聚糖溶液濃度0.86%,初次凝膠時間30.71 min,該工藝制備微球的平均粒徑為329 nm,且呈正態分布,藥物包封率為94.09%。
猜你喜歡
河北科技師范學院學報(2022年2期)2022-08-26 08:55:40
河北科技師范學院學報(2021年1期)2021-05-10 03:34:20
中成藥(2017年12期)2018-01-19 02:06:57
電源技術(2017年1期)2017-03-20 13:37:59
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
天然產物研究與開發(2016年1期)2016-06-05 10:29:25
食品界(2016年4期)2016-02-27 07:36:46
中國果菜(2015年2期)2015-03-11 20:01:01
應用化工(2014年7期)2014-08-09 09:20:21
應用技術學報(2014年4期)2014-02-28 14:52:40
