超高效液相色譜-串聯(lián)質(zhì)譜法快速測定保健食品中大豆異黃酮含量
2019-09-11 07:55:08胡珀,金華
食品工業(yè)科技 2019年13期
關鍵詞:大豆
胡 珀,金 華
(淮安市食品藥品檢驗所,江蘇淮安 223001)
大豆異黃酮是大豆中的一類次生代謝產(chǎn)物,屬黃酮類中的異黃酮多酚化合物[1]。目前發(fā)現(xiàn)的大豆異黃酮共有12種,包括游離型苷元和相應糖苷[2]。大豆異黃酮是天然的優(yōu)質(zhì)抗氧化劑,具有很強的清除羥自由基(-OH)的能力;具有防癌抗腫瘤、降低膽固醇、預防骨質(zhì)疏松和心血管疾病、延緩衰老、改善婦女更年期綜合癥等多種生理功能[3-6]。因此,以大豆異黃酮為主要活性成分的保健食品的開發(fā)和應用受到廣泛關注。其中大豆苷(Daidzin)、大豆黃苷(Glycitin)、染料木苷(Genistin)、大豆素(Daidzein)、大豆黃素(Glycitein)以及染料木素(Genistein)六種大豆異黃酮單體是大豆中異黃酮的主要組分。
目前,保健食品市場存在生產(chǎn)低水平重復、價位較高、夸大產(chǎn)品功效的現(xiàn)象。因此,應該提高保健食品功效成分的檢測分析水平[7]。保健食品中大豆異黃酮含量的傳統(tǒng)測定方法有紫外分光光度法(UV),該法具有方法簡便、重現(xiàn)性好等優(yōu)點,但特異性較差,無法判斷樣品中各組分的構成情況[8];薄層掃描法(TLCS),該法具有取樣量少、分離效果好等優(yōu)點,能較好地進行定性分析,但定量檢測精度不高,人為誤差較大[9];氣相色譜法(GC)法具有進樣量少、特異性高等優(yōu)點,但在測定時需制備衍生物,步驟較多,耗時長,從而限制了該法的推廣應用[8];酶聯(lián)免疫吸附法(ELISA)具有檢測靈敏度高、檢測速度快的優(yōu)點,多用于生物體血液及尿樣中大豆異黃酮的檢測[10];高效液相色譜法(HPLC)主要是參照GB/T 23788-2009《保健食品中大豆異黃酮的測定方法高效液相色譜法》[11],該分析方法具有自動化程度高、測定結果準確、重現(xiàn)性好等優(yōu)點,但儀器運行時間較長,需要60 min,不適合研發(fā)檢測,另外,待檢樣品基質(zhì)復雜,大豆異黃酮含量低,不易檢出,長期直接進樣,易污染色譜柱。
本課題采用固相萃取-超高效液相色譜-串聯(lián)質(zhì)譜的方法對保健食品中的大豆異黃酮進行檢測,采用固相萃取技術,去除了油性基質(zhì)干擾;采用超高效液相色譜分離技術,增加分析的通量,縮短了分析時間,同時減少溶劑用量,降低分析成本[12];通過質(zhì)譜檢測器檢測,實現(xiàn)高靈敏度,檢出濃度達到納克數(shù)量級。研究結果表明,此種方法樣品前處理簡單、有機試劑消耗少、時間短,能夠滿足大豆異黃酮定性和定量檢測需求,為保健食品的質(zhì)量評價及研究提供有價值的參考。
1 材料與方法
1.1 材料與儀器
大豆磷脂軟膠囊 湯臣倍健股份有限公司,批號20180401G;倍仕好牌大豆異黃酮軟膠囊 浙江康恩貝健康科技有限公司 批號R1810021;百合康牌大豆卵磷脂軟膠囊 威海百合生物技術股份有限公司 批號412688012;大豆苷、大豆黃苷、染料木苷、大豆素、大豆黃素以及染料木素(純度均≥90%) 上海安譜實驗科技有限公司;甲醇、乙腈(色譜級) 德國Merker公司;甲酸(色譜級) 德國CNW公司;CNWBOND Si、CNWBOND Florisil、CNWBOND HC-C18、CNWBOND LC-C18、Poly-Sery MCX固相萃取柱 上海安譜實驗科技有限公司;實驗用水由Milli-Q純水機制得。
XS205DU電子天平 瑞士METTLER TOLEDO公司;API 4000+三重四級桿串聯(lián)質(zhì)譜儀 美國AB公司;H-CLASS超高效液相色譜儀 美國Waters公司;2695/2998高效液相色譜儀 美國Waters公司;KH-100B型超聲波清洗器 昆山禾創(chuàng)超聲儀器有限公司;X1R型高速低溫離心機 美國ThermoFisher公司;Milli-Q超純水機 美國Millipore公司。
1.2 實驗方法
1.2.1 樣品制備 取20粒軟膠囊樣品,傾出內(nèi)容物,用干凈玻璃棒攪拌均勻,現(xiàn)用現(xiàn)制。
1.2.2 標準儲備溶液的配制 分別稱取大豆苷、大豆黃苷、染料木苷、大豆素、大豆黃素以及染料木素標準品0.0100 g于100 mL容量瓶中,以甲醇定容至刻度,得100 μg/mL大豆苷、大豆黃苷、染料木苷、大豆素、大豆黃素以及染料木素單標儲備溶液,4 ℃避光保存。
1.2.3 混合標準使用溶液的配制 分別移取1 mL大豆苷、大豆黃苷、染料木苷、大豆素、大豆黃素以及染料木素單標儲備溶液于同一100 mL容量瓶中,用甲醇定容得濃度為1 μg/mL混合標準中間溶液。分別移取0.01、0.02、0.05、0.1、0.2、0.5、1、2、5 mL混合標準中間溶液于10 mL容量瓶中,用甲醇定容至刻度,得濃度為1、2、5、10、20、50、100、200、500 μg/L的混合標準使用溶液,以此繪制標準曲線,外標法定量。
1.2.4 樣品處理 稱取0.5 g樣品置于50 mL容量瓶中,加80%甲醇溶液至接近刻度,超聲提取20 min,用80%甲醇溶液定容,搖勻。取樣品溶液置于離心管中,8000 r/min離心10 min。取上清液1 mL全部通過CNWBOND Florisil固相萃取柱(預先以5 mL甲醇,5 mL水活化),以5 mL水淋洗柱體,25 MPa加壓抽干5 min。以6 mL甲醇洗脫柱體,于40 ℃氮氣吹干,以1 mL 80%甲醇溶液溶解,過0.22 μm微孔濾膜后待測定。
1.2.5 高效液相色譜串聯(lián)質(zhì)譜條件 色譜柱:Waters ACQUITY UPLC C18柱(2.1 mm×100 mm,1.7 μm);流動相:A相為0.1%甲酸水溶液,B相為乙腈;梯度洗脫程序為:88% A 0~4.0 min;88%~80% A 4.0~6.0 min;80% A 6.0~8.0 min;80%~70% A 8.0~10.0 min;70%~20% A 10.0~10.1 min;20% A 10.1~12 min,流速0.3 mL/min,柱溫30 ℃,進樣體積2 μL。質(zhì)譜離子源為ESI(+),霧化電壓:5500 V;離子傳輸溫度:500 ℃,各大豆異黃酮多反應監(jiān)測(MRM)模式監(jiān)測參數(shù),見表1。
1.2.6 定性、定量檢測
1.2.6.1 定性檢測 取1.2.3 中50 μg/L的混合標準使用溶液和1.2.4中樣品溶液按色譜條件1.2.5進行測定,依據(jù)標準品色譜峰保留時間,對樣品溶液中的組分進行定性,保留時間應一致。
1.2.6.2 定量檢測 將1.2.3中大豆異黃酮混合標準使用溶液在色譜條件1.2.5下進行測定,繪制以峰面積為縱坐標、濃度為橫坐標的標準曲線。將1.2.4制備的樣品溶液注入液質(zhì)聯(lián)用儀中,保證樣品溶液中大豆苷、大豆黃苷、染料木苷、大豆素、大豆黃素以及染料木素的響應值均在工作曲線的線性范圍內(nèi),若超出最高濃度,將樣品稀釋后再進樣。由標準曲線得到樣品溶液中大豆苷、大豆黃苷、染料木苷、大豆素、大豆黃素以及染料木素的濃度。
2 結果與分析
2.1 固相萃取柱的選擇
樣品未經(jīng)過除油處理,會殘留較多油溶性雜質(zhì),如亞油酸、油酸等,特別是批量進樣后,色譜柱受污染更重,致使系統(tǒng)壓力明顯升高,很容易超過警戒最高壓力[13]。
通過固相萃取能夠去除大豆苷之前的大部分雜峰,很好地降低了樣液對液相色譜柱的污染損耗,延長了色譜柱的使用時間[14]。實驗考察了5種不同填料的固相萃取柱,測定樣品在添加1 mL 50 μg/L大豆異黃酮混合標準使用溶液的回收率。表2結果表明Florisil固相萃取柱有較好的回收率和凈化效果,大豆異黃酮中各組分回收率最高,平均回收率為85.4%,并具有污染小、可處理小體積試樣等優(yōu)點,能夠在保留目標化合物的同時,除去油性樣品中的干擾物,可用于大豆異黃酮分離純化。Florisil 固相萃取柱中的萃取介質(zhì)弗羅里硅土作為氧化鎂復合的極佳硅膠吸附劑,適合于從非極性基質(zhì)中吸附極性化合物,具有高吸附容量、高靈敏度、方便耐用和穩(wěn)定等優(yōu)點[15]。
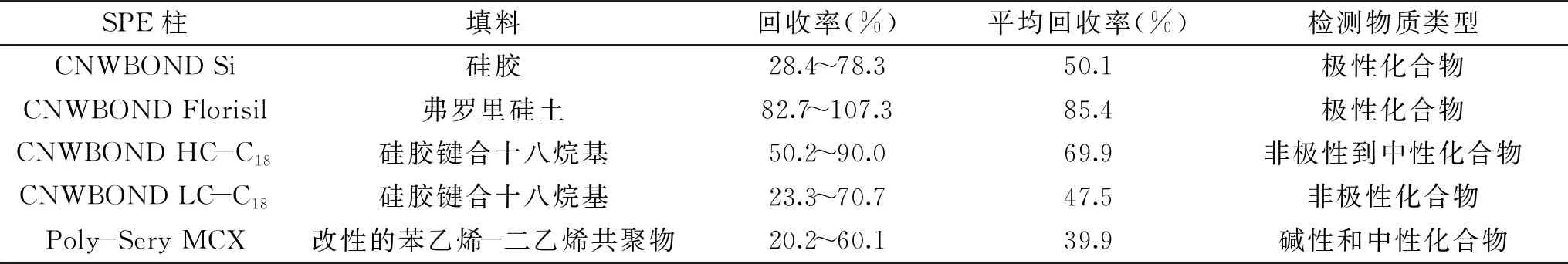
表2 不同固相萃取柱(SPE)的回收率實驗結果Table 2 Experimental results on recovery of different solid phase extraction columns(SPE)
2.2 質(zhì)譜條件的選擇
大豆異黃酮各組分在質(zhì)譜上具有很強的正離子響應,因此選擇正離子模式進行掃描。500 μg/L混合標準使用溶液采用蠕動泵進樣分析,在正離子模式下,6種大豆異黃酮各組分一級質(zhì)譜均出現(xiàn)[M+H]+準分子離子峰,且豐度較大,因此選擇分子離子峰作為母離子。分別對6中大豆異黃酮各組分母離子進行轟擊碎裂,得到特征碎片離子峰信息,確定各目標物的特征子離子,具體見表1。在MRM模式下優(yōu)化碰撞能量、去簇電壓,確定最佳質(zhì)譜檢測條件。
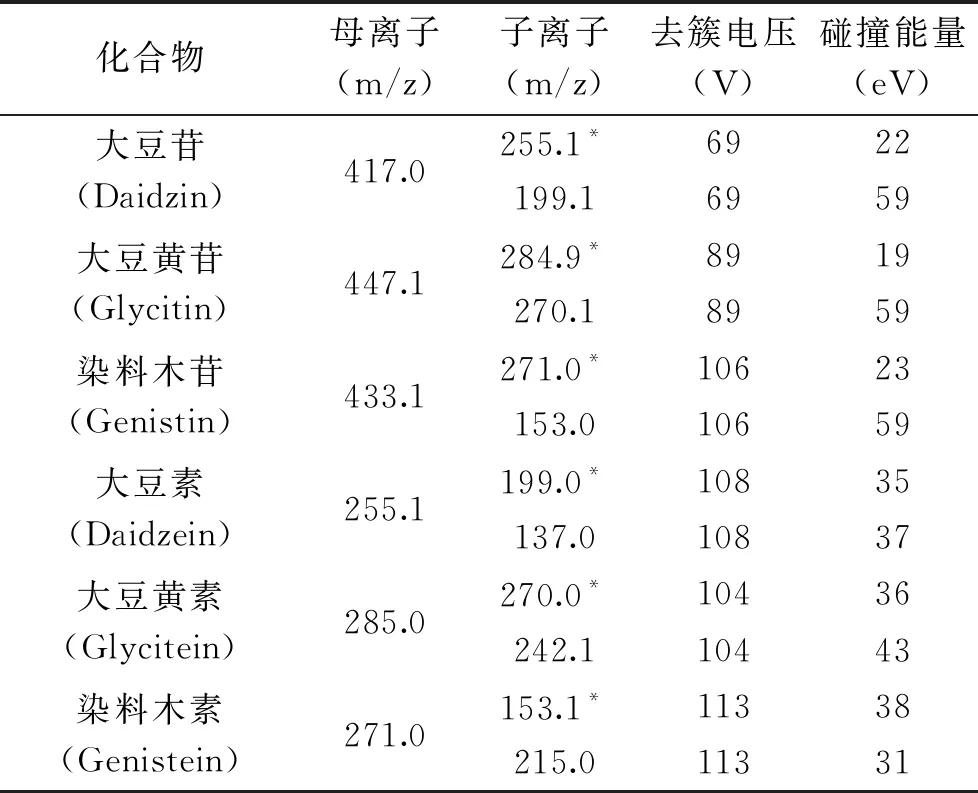
表1 6種大豆異黃酮質(zhì)譜分析參數(shù)Table 1 MS/MS parameters of 6 soybean isoflavones
2.3 色譜條件的選擇
2.3.1 色譜柱的選擇 以C18色譜柱作為分析柱。通過系列實驗,比較了Waters ACQUITY UPLC C18柱(2.1 mm×100 mm,1.7 μm)、Agilent Eclipse Plus C18(2.1 mm×50 mm,1.8 μm)和Agilent Eclipse XDB-C18(2.1 mm×50 mm,1.8 μm)三種色譜柱對6種大豆異黃酮各組分的分離效果。結果表明,在相同色譜條件下Waters ACQUITY UPLC C18柱對異黃酮的分離效果最好,能提供良好的峰形,因此選擇該色譜柱作為分析柱。
2.3.2 流動相的選擇 考察了不同流動相對異黃酮色譜行為的影響。以含乙腈-水、甲醇-水作流動相,在采取梯度洗脫的方式時,大豆苷和大豆黃苷沒有完全分離開;采用0.5%的乙酸水溶液和甲醇作為流動相,大豆素和大豆黃素沒有完全分離開;采用20 mmol/L乙酸銨水溶液和乙腈作為流動相,大豆黃素與染料木素色譜峰重合;以0.1%甲酸水溶液-乙腈為流動相,通過優(yōu)化兩相的配比,6種異黃酮均可獲得滿意的分離度。通過優(yōu)化梯度洗脫條件,獲得了6個目標物合適的保留時間和峰形,6種大豆異黃酮總離子流圖見圖1。

圖1 6種大豆異黃酮總離子流圖Fig.1 Total ion chromatogram of 6 kinds of soybean isoflavones注:1:大豆苷;2:大豆黃苷;3:染料木苷; 4:大豆素;5:大豆黃素;6:染料木素。
2.4 方法的線性關系與檢出限
分別將1.2.3的混合標準使用溶液按色譜條件進行測定,以6種大豆異黃酮各組分的色譜峰面積(y)和質(zhì)量濃度(x)做線性回歸,結果顯示大豆苷、染料木苷、大豆素以及染料木素在10~500 μg/L范圍內(nèi),大豆黃苷在5~500 μg/L范圍內(nèi)、大豆黃素在1~200 μg/L范圍內(nèi)線性關系均良好(r>0.99),見表3。以3倍的信噪比確定方法的檢出限,大豆黃苷、大豆黃素檢出限均為10 μg/kg,定量限均為30 μg/kg,大豆苷、染料木苷檢出限均為20 μg/kg,定量限均為60 μg/kg,大豆素、染料木素檢出限均為 30 μg/kg,定量限均為90 μg/kg。
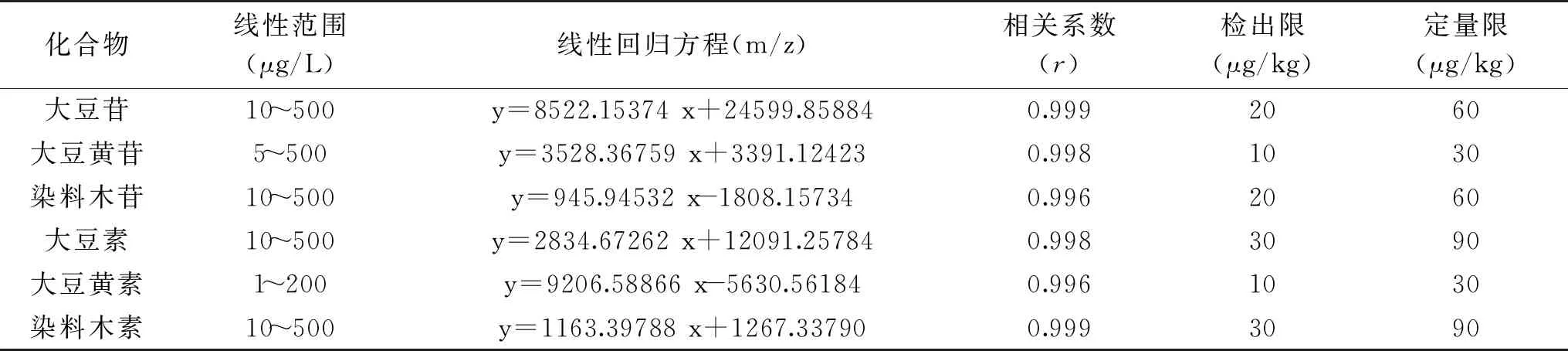
表3 6種大豆異黃酮各組分的線性關系與檢出限Table 3 Linear relationship and detection limit for 6 soybean isoflavones
2.5 方法的回收率和精密度考察
在上述優(yōu)化后的試驗條件下,分別對樣品添加2、5和10 mg/kg的標準品后進行測定,平均回收率和精密度見表4。結果顯示,加標回收率為81.8%~98.4%,相對標準偏差為1.8%~6.7%,能夠滿足檢測方法的要求。
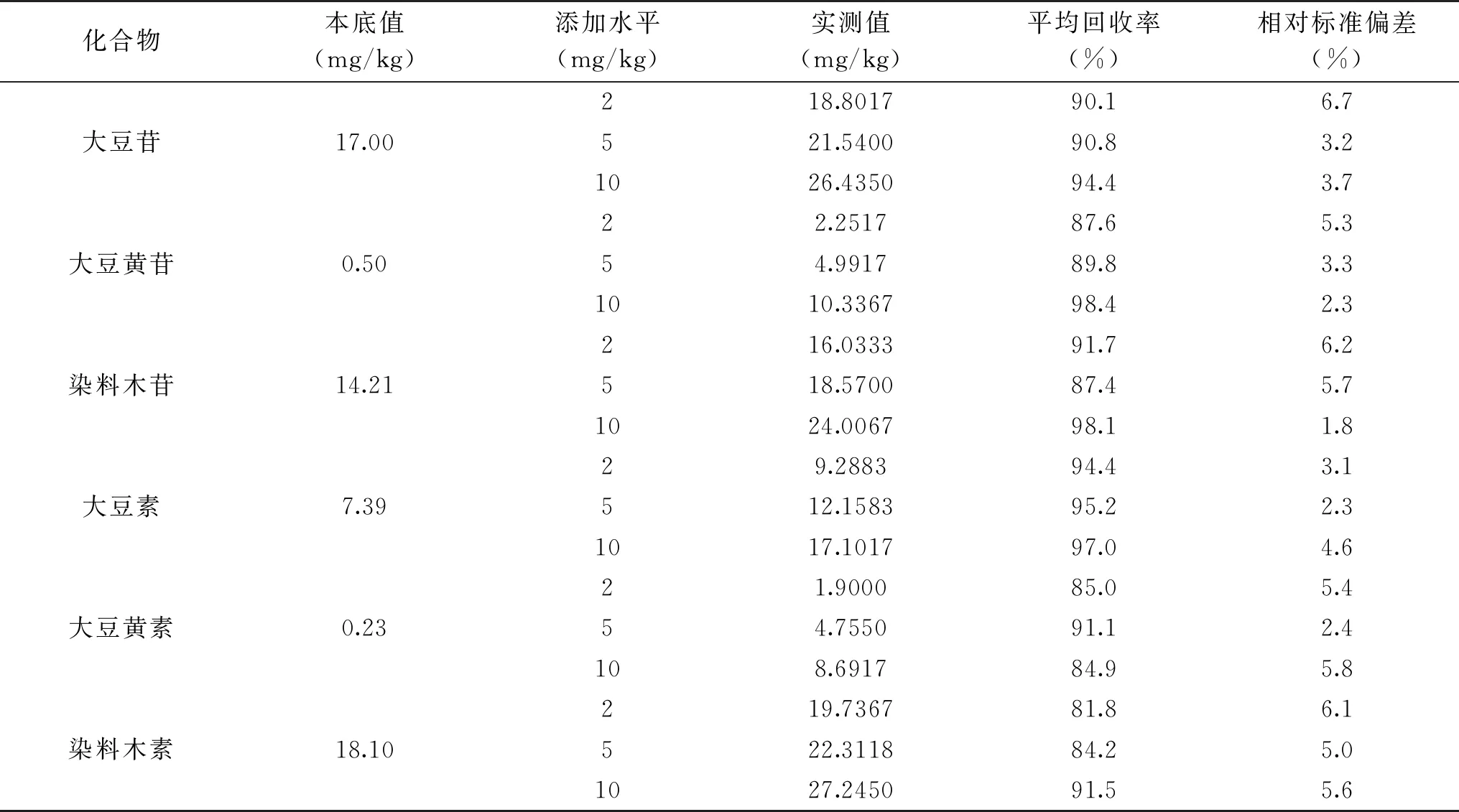
表4 樣品中6種大豆異黃酮各組分的加標回收率和精密度(n=6)Table 4 Results of recovery and repeatability for 6 soybean isoflavones(n=6)
2.6 實際樣品中6種大豆異黃酮各組分檢測
取湯臣倍健大豆磷脂軟膠囊、倍仕好牌大豆異黃酮軟膠囊、百合康牌大豆卵磷脂軟膠囊,按照標準GB/T 23788-2009[11]和本文方法進行檢測,樣品中大豆異黃酮含量的測定結果見表5。結果表明,標準GB/T 23788-2009方法檢出限為5 mg/kg,樣品中的大豆苷、大豆黃素若含量低于5 mg/kg,則未檢出,而且在進樣過程中,系統(tǒng)壓力過大,兩次報錯,說明前處理方法不合理,未經(jīng)去油處理直接進樣會堵塞色譜柱,而超高效液相色譜串聯(lián)質(zhì)譜檢出限為10~30 μg/kg,低含量的物質(zhì)也可以精確定量,通過Florisil固相萃取柱凈化,去除了油性基質(zhì)干擾,整個進樣過程中,系統(tǒng)壓力穩(wěn)定。兩種方法的檢測結果偏差在2.2%~8.3%,結果可信。
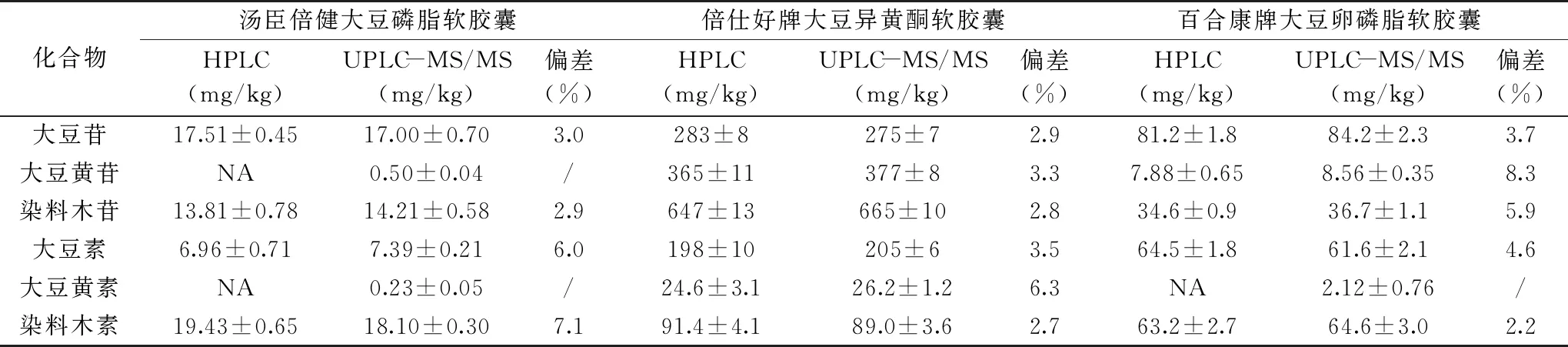
表5 樣品中大豆異黃酮含量的測定結果Table 5 Determination results of soybean isoflavones contents in the samples
3 結論
建立了保健食品中大豆苷、大豆黃苷、染料木苷、大豆素、大豆黃素以及染料木素超高效液相色譜串聯(lián)質(zhì)譜同時檢測方法。結果表明,大豆苷、大豆黃苷、染料木苷、大豆素、大豆黃素以及染料木素在各自濃度范圍內(nèi)線性關系良好;大豆黃苷、大豆黃素檢出限均為10 μg/kg;大豆苷、染料木苷檢出限均為20 μg/kg;大豆素、染料木素檢出限均為30 μg/kg,加標回收率為81.8%~98.4%,相對標準偏差為1.8%~6.7%。通過與標準GB/T 23788-2009液相方法結果比較,該方法具有較高的靈敏度和較低的檢出限,在實際檢測工作中具有良好應用價值。
猜你喜歡
農(nóng)業(yè)科技通訊(2023年1期)2023-02-12 07:09:18
今日農(nóng)業(yè)(2022年16期)2022-11-09 23:18:44
中國化肥信息(2022年7期)2022-08-31 01:29:28
中國化肥信息(2022年5期)2022-08-30 01:58:26
今日農(nóng)業(yè)(2021年20期)2021-11-26 01:23:56
今日農(nóng)業(yè)(2021年14期)2021-10-14 08:35:34
下一代英才(酷炫少年)(2018年6期)2018-07-09 03:17:44
農(nóng)產(chǎn)品市場周刊(2017年4期)2017-03-03 19:40:05
兒童故事畫報·智力大王(2015年10期)2016-01-27 01:01:35
讀寫算(中)(2015年10期)2015-11-07 07:24:12
